Updates to the publicly available 10x Genomics transcriptome and V(D)J references are documented separately.
Enhancements and updates
- Cell Ranger v9.0.1 is bundled with mouse probe set reference v1.1.1.
System requirements
- AVX instructions must be available on CPUs which running Cell Ranger v9.0.0 or higher. AVX instructions were introduced in 2011 on both Intel and AMD CPUs. In the future, AVX2 will be required. AVX2 instructions were introduced in 2013 on Intel CPUs and in 2015 on AMD CPUs. The System Requirements page has been updated accordingly.
Bug fixes
- Fixed a bug which led to analysis failures on CPUs that lack AVX2 support.
- Disabled numpy CPU dispatch for AVX-512. This eliminates a source of minor variation in results between machines with and without AVX-512 available that could occur in v9.0.0.
- Fixed a bug impacting sitecheck when the filesystem is not accessible. Sitecheck no longer uses
statfson all mounted systems. - Fixed a bug that prevented
cellranger vdjfrom processing paired end GEM-X VDJ libraries. With this fix, bothcellranger multiandcellranger vdjcan be used to analyze VDJ-only libraries. - GEM-X 3’ Universal Gene Expression + CRISPR analysis has been enabled.
- Resolved an issue in
cellranger aggrwhere web summary UMAP cluster annotations were incorrectly plotted for cell number greater than 10,000, and the UMAP coordinates appeared identical to the first sample listed in the aggregation CSV. - Fixed a bug in
cellranger mkrefthat caused an error message to appear even after a successful run. - Fixed a bug that caused the analysis pipeline to crash when the experimental design included hashing with Antibody Capture for a single feature.
Major updates
- GEM-X Universal on-chip multiplexing (OCM) support: Cell Ranger v9.0 now supports the analysis of GEM-X Universal 3'v4 and 5'v3 Gene Expression 4-plex assays, enabling the analysis of on-chip multiplexed samples via read-based demultiplexing.
- GEM-X Flex libraries support: Cell Ranger v9.0 now supports the analysis of GEM-X Flex libraries.
- Telemetry data collection: Cell Ranger v9.0 includes anonymized telemetry data collection to help 10x Genomics improve product functionality. This data provides insights into usage patterns, aids in diagnosing issues, and helps prioritize feature improvements. For more information on the collected data and its usage, please visit https://10xgen.com/pipeline-telemetry.
- Antibody hashtag analysis enabled: Cell Ranger v9.0 enables the ability to analyze samples multiplexed using antibody hashtags (also known as cell or sample hashing), allowing
hashtag_idsto be specified in themulti config CSVto demultiplex antibody count data similarly to CellPlex oligos (CMOs). This process utilizes the JIBES algorithm to assign antibody hashtags to cell barcodes and extends compatibility for demultiplexing in both 3' and 5' Gene Expression workflows, including V(D)J analysis. Antibody-based hashing is not compatible with OCM. Antibody hashtag analysis is not officially supported. - Redesigned web summary HTML: Cell Ranger v9.0 features a redesigned web summary HTML, with improved navigation that makes it easier to find key data quality metrics.
- Flex renaming: Fixed RNA profiling has been renamed Flex. All future updates, documentation, and support will reference Flex.
cellranger mkfastqdeprecation: Thecellranger mkfastqpipeline is now deprecated and will be removed in the next Cell Ranger release.- Automated cell type annotations: Cell Ranger v9.0 introduces the ability to perform automated cell type annotations. This feature is available as part of
cellranger countandcellranger multior as a standalone command,cellranger annotate. Currently only available for human samples, the model is a beta feature.- A 10x Genomics Cloud Analysis account is required to perform automated cell type annotations, as the process is conducted within Cloud Analysis.
- Users must agree to the 10x Genomics End User License Agreement (EULA) before utilizing this feature.
- A new command,
cellranger cloud auth, has been introduced to simplify the retrieval of the cloud account security token, a required input for performing automated cell type annotations. - Learn about the new output files generated to support cell type annotations.
General improvements
- Cell Ranger v9.0 now outputs UMAP projections instead of t-SNE in
cellranger aggrto improve processing speed. In internal testing, UMAP was 2.5 times faster than t-SNE, significantly reducing runtime, especially for larger datasets.- An optional argument,
--enable-tsnehas been added tocellranger aggrfor users who wish to include t-SNE projections in the.cloupeandweb_summary.html. This option defaults to false. - The
cellranger multipipeline also defaults to UMAP. However, t-SNE is still calculated and available for visualization in the.cloupefile. The--enable-tsneargument is not applicable tomulti.
- An optional argument,
- General performance improvements resulting in faster analysis times across workflows.
- Improved the error message to indicate that FASTQ files with the same name are located in different subdirectories.
Universal 3’ and 5’ Single Cell Gene Expression
- The
cellranger multipipeline now supports autodetection of paired-end (PE) VDJ libraries. This feature is not yet available incellranger vdj. - Enclone deprecation: In Cell Ranger v5.0 to v8.0, clonotype grouping was performed using enclone, which filtered and grouped cells into clonotypes. Starting with Cell Ranger v9.0, clonotype grouping and filtering are now fully integrated within Cell Ranger. As a result, enclone has been deprecated.
- VDJ can now be specified as a
library_typefor multiple FASTQ sets. The algorithm automatically detects whether the FASTQ files correspond to TCR or BCR libraries, streamlining the analysis process. - The analysis of standalone Antibody Capture libraries (without Gene Expression) no longer requires a transcriptome reference as an input in
cellranger countandcellranger multi. - Resolve the GTF file parsing issue where an exon entry appears before its corresponding gene entry, which previously led to the error message:
transcript FOO is missing. - In general, the analysis of VDJ-only libraries (without an accompanying Gene Expression library) is enabled and supported. However, with OCM, the analysis of VDJ-only libraries is enabled but unsupported.
Algorithm changes
- The EmptyDrops false discovery rate (FDR) threshold has been lowered to 0.001 for 3' and 5' analyses. This adjustment may lead to a decrease in the number of cells called, improving the detection sensitivity in these analyses.
- Fixed a rare bug in the EmptyDrops cell calling algorithm that could result in no cells being called.
- VDJ algorithm updates
- Ensures that V and J genes from different chains are no longer allowed to be productive. This change improves the accuracy of clonotype assignments.
- Fixed an issue where CDR3 sequences located outside the V-J region were incorrectly marked as productive. Although downstream filtering excluded these sequences from clonotypes, this annotation error has now been corrected to ensure that only CDR3 sequences within the V-J region are marked as productive.
- A new chimera filter has been introduced that removes barcodes with two contigs sharing the same V region but with different CDR3 sequences within the same barcode. This filter improves clonotype accuracy by excluding barcodes likely representing chimeric artifacts.
- Removed logic that based TRBC1/2 annotation on J-REGION annotation, simplifying and improving the accuracy of T cell receptor (TCR) annotations. This change ensures TRBC1/2 annotations are assigned independently of J-REGION annotations, reducing dependency on specific gene region annotations.
- Added a filter for barcodes with chimeric contigs that share the same V-REGION but have different CDR3 sequences. This update improves clonotype assignment.
- Introduced a filter to remove contaminating barcodes caused by gel bead indels. Pairs of barcodes with identical productive contigs are compared based on UMI counts. When one barcode (source) has a UMI count at least 10 times higher than the other (sink), and the sink barcode sequence differs from the source by a 1 bp indel, the sink barcode is marked as a contaminant and excluded.
Output files
- Web summary update:
- The "Top 10 Clonotypes" table in the web summary for VDJ libraries has updated column labels: "Frequency" has been changed to "Number of Cells," and "Proportion" is now labeled "Fraction of Cells."
- Cell Ranger v9.0 addresses an issue where the median IGL UMI metric appeared in the web summary for samples with only IGH/IGK clonotypes, even when all IGL contigs were unproductive. The web summary now accurately reflects the filtered, high-quality contig data, and unproductive IGL contigs are excluded from hero metrics calculations.
- VDJ Median UMIs metric now excludes UMIs from unproductive contigs in computation.
- The contig annotation output files for 5' Immune Profiling experiments now include a new "Sample" column. This column remains blank for singleplex VDJ libraries run through
cellranger vdj. Additionally, theraw_clonotype_idcolumn is now prefixed with the clonotype ID assigned to each corresponding cell barcode. - Added a
productive_criteriafield to theall_contig_annotations.jsonfile. This field includes a boolean value for each of seven criteria required for a contig to be labeled as productive. To be considered productive, a contig must meet all seven criteria:
"productive_criteria": {
"full_length": true,
"has_v_start": true,
"in_frame": true,
"no_premature_stop": true,
"has_cdr3": true,
"has_expected_size": true,
"correct_ann_order": true
}
Flex
- Cell Ranger v9.0 is bundled with the newly released 2024-A probe set reference for human and mouse. Genes not present in the 2024-A reference but included in the older 2020-A reference are now excluded from the analysis. This update also resolves a bug where an excluded gene, missing from the transcriptome reference, was mistakenly included in the filtered feature-barcode matrix. Previously, the missing gene was incorrectly identified as an exogenous gene (e.g., GFP), which should be retained in the filtered matrix.
- For the Human GRCh38-2024-A reference:
- 99 probes targeting 83 genes have been added back, as they are now recognized as target-specific.
- 84 probes targeting 33 genes have been excluded and removed from GRCh38-2024-A.
- For the Mouse GRCm39-2024-A reference:
- 32 probes targeting 29 genes have been added back, as they are now recognized as target-specific.
- 41 probes targeting 18 genes have been excluded and removed from GRCm39-2024-A.
- For the Human GRCh38-2024-A reference:
- Transcriptome reference is now an optional input file for Flex libraries. If the transcriptome reference is omitted and
create-bam = true, the pipeline generates unaligned BAM output files. - New optional parameter
emptydrops-minimum-umisto manually specify the UMI cutoff during the second step of cell calling. See https://10xgen.com/scFFPE-cell-calling for details. - The descriptions of Flex chemistry options have been updated to reflect the renaming of "Fixed RNA Profiling" to "Flex," for consistent terminology across documentation and interfaces.
Algorithm changes
- The EmptyDrops background range for sub-pooled samples has been updated from 22,500–45,000 to 45,000–90,000 per multiplexing barcode based on experimetnal data.
Known bugs
- Cell Ranger v9.0.0 cannot process GEM-X 3’ Universal Gene Expression + CRISPR libraries.
- In v9.0.0, GEM-X paired end VDJ-only analysis is supported exclusively in the
multipipeline. It is not available in thevdjpipeline. - The Mouse Probe Set v1.1.0 differs in two gene names when compared to the GRCm39-2024-A transcriptome reference. These discrepancies have been resolved in Mouse Probe Set v1.1.1, which is now bundled with Cell Ranger v9.0.1. If you are using Cell Ranger v9.0.0, please download the Mouse Probe Set v1.1.1 separately.
New input parameters
- The
SCP5-PE-v3chemistry option has been added to the cellranger count and cellranger multi pipelines. This addition allows for the analysis of Single Cell 5′ paired-end v3 (GEM-X) libraries, where both R1 and R2 reads are utilized for alignment. - A new optional parameter,
min-crispr-umi, has been introduced in the cellranger count and cellranger multi pipelines. This parameter allows users to customize the minimum number of CRISPR guide RNA UMIs required for protospacer detection, enabling adjustments in detection sensitivity based on experimental needs. The recommended default threshold is 3 guide RNA UMIs.
Minor algorithm changes and bug fixes
- The EmptyDrops background range for NextGEM HT chemistries (
SC3Pv3HTandSC5PHT) has changed from 45,000-90,000 to 80,000-160,000 to optimize cell calling. This change addresses undercalling issues noted in v8.0.0 for HT cell loads over 30,000 cells. When analyzing NextGEM HT libraries in v8.0.1 and later, users must manually specifySC3Pv3HTorSC5PHTchemistry. This updated background range will not apply when chemistry is autodetected. - Fixed a bug in the
--force-cellsparameter that previously caused an error (during indexing in the FILTER_BARCODES stage) when the number of specified cells matched the number of barcodes. - Fixed a bug where references containing genes without transcripts lead to the incorrect selection of genes in the filtered feature-barcode matrix.
New features
-
Cell Ranger v8.0 introduces 3’ v4 and 5’ v3 chemistry to support the analysis of Chromium GEM-X Single Cell Gene Expression v4 and Chromium GEM-X Single Cell Immune Profiling v3 libraries.
-
Cell Ranger v8.0 introduces support for protein labeling with Proteintech Genomics (PTG)-derived antibodies. This addition allows researchers to simultaneously analyze intracellular and extracellular protein expression within single cells. Antibody Capture libraries may be a mix of BioLegend and PTG antibody-labeled cells. For detailed instructions on setting up your assay, please refer to this Demonstrated Protocol.
-
Cell Ranger v8.0 enables the analysis of Flex CRISPR Guide Capture libraries, providing the ability to analyze the transcriptome from fixed cells that have undergone CRISPR-based perturbations. However, this is an unsupported workflow, meaning it is not officially supported by technical support and may have limitations.
- For guidance on setting up the analysis, please refer to this example multi config CSV.
- For instructions on designing probes for CRISPR Guide Capture with the Flex assay, please refer to this Knowledge Base article.
- Barcode auto-pairing is disabled for CRISPR Probe Barcodes.
Command line changes
-
The optional command line parameter
–no-bam, applicable to thecellranger countandcellranger multipipelines, has been replaced with a new required parameter called –create-bam. -
Chemistry names have been revised for some Flex datasets. New chemistry names are:
SFRP: Flex (Singleplex)MFRP: Flex (Multiplex, Probe Barcode on R2)MFRP-R1: Flex (Multiplex, Probe Barcode on R1)MFRP-RNA: Flex (Multiplex, RNA, Probe Barcode on R2)MFRP-Ab: Flex (Multiplex, Antibody, Probe Barcode at R2:69)MFRP-Ab-R2pos50: Flex (Multiplex, Antibody, Probe Barcode at R2:50)MFRP-RNA-R1: Flex (Multiplex, RNA, Probe Barcode on R1)MFRP-Ab-R1: Flex (Multiplex, Antibody, Probe Barcode on R1)
-
A new column called 'chemistry' has been added to the library table in the multi config CSV for specifying the chemistry of each library separately. Currently, this is only relevant for Flex data analysis.
Outputs
-
The output file
donor_regions.fafor 5' V(D)J libraries generated bycellranger multihas been relocated. Previously found at~/outs/reference/donor_regions.fa, it now resides in~/outs/per_sample_outs/donor_regions.fa. -
Similarly, the
donor_regions.fafile produced bycellranger vdjhas moved from~/outs/vdj_reference/fasta/donor_regions.fato the main~/outs/directory.
Algorithm updates & improvements
-
The chemistry detection algorithm has been updated to subsample 100,000 reads from the first 2 million reads (previously subsampled from the first 1 million reads).
-
Cell calling algorithm updates:
- The EmptyDrops background range was increased for 3’ v4 and 5’ v3 inclusion list data to 80k-160k.
- Minimum UMI threshold of Empty Drops was changed to:
max(500, 1+max UMI observed in the ambient range). This change ensures more accurate classification, although some barcodes that previously passed the filter may now be classified as background. - Previously, Cell Ranger’s cell calling algorithm included a filtering step based on the median of OrdMag. Our evaluation of internal data and customer feedback revealed that this EmptyDrops threshold was overly stringent, resulting in the excessive removal of cell barcodes from high UMI samples. To improve the recovery of low RNA cell barcodes distinct from background, we removed this restriction in Cell Ranger v8.0.0 and later versions. As a result, some customers may now recover more cells. Learn more about Cell Ranger's cell calling algorithm.
-
CRISPR Guide Capture algorithm updates:
- For enhanced guide RNA detection accuracy, a cell barcode now requires a minimum of three guide RNA UMIs to be considered positive for that guide RNA. This update aims to reduce false positives, ensuring more reliable CRISPR analysis results.
- The guide RNA detection (protospacer calling) algorithm's runtime has been substantially improved.
Deprecated and unsupported features
-
Support for Low Throughput (LT) libraries has been disabled. Use Cell Ranger v7.2 to analyze your LT libraries.
-
cellranger aggrno longer supports the aggregation of Targeted Gene Expression. -
cellranger aggrno longer supportsmolecule_info.h5v2 and older. These files are typically generated by Cell Ranger v2.2 and earlier. -
Ubuntu 14.04 and Westmere CPUs are no longer supported.
Minor updates and bug fixes
-
The interactive features of the
cellranger countandcellranger multibarcode rank plot have been enhanced. Now, hovering over a region of the plot reveals detailed information, including the total number and percentage of barcodes (in that region) identified as cells, UMI counts for these barcodes, and their barcode ranks, sorted by UMI counts in descending order. -
The
cellranger countweb summary has been updated to include a 'Command Line Arguments' section in the Summary tab. The complete command used to run thecellranger countprocess is displayed. This enhancement aids users in tracking and documenting the specific parameters used during the analysis, facilitating reproducibility and troubleshooting. -
Intron mode informational alert in the web summary HTML output has been removed.
-
A bug has been fixed to ensure the
--jobmode clusterargument is now properly recognized and applied. -
Some errors that prevented V(D)J clonotype analysis from completing (error messages containing ‘RUST PANIC ERROR’) have been resolved.
Known issues
-
High-Throughput (HT) runs with Cell Multiplexing: For >30,000 cells, a reduction of up to 10% in the number of cells detected may occur. Customers experiencing this issue should continue with Cell Ranger v7.2 for optimal results. To upgrade to Cell Ranger v8.0 and still achieve the expected cell counts in high-plex samples, use the
--force-cellsoption with the anticipated number of cells. -
The 2024-A mouse transcriptome reference uses the new coordinate system GRCm39.
-
Web summary aggregate filter display issue: When disabled, the filter incorrectly displays ‘Antibody Aggregate Filter’
<False>across Gene Expression and Antigen summaries. It should be labeled ‘Aggregate Filter’ and only appear for Antibody and Antigen data. This issue will be addressed in the upcoming release of Cell Ranger.
New feature: Flex with multiplex Antibody Capture
-
Cell Ranger v7.2 is required for analysis of Flex data with multiplexed Gene Expression and Antibody Capture libraries. Instructions for running the
cellranger multisubcommand are described in the running multi pipeline page. Output files are described in the Understanding Outputs section. The Flex algorithms section includes descriptions of the new methods that were developed for processing multiplexed Gene Expression and Antibody Capture data. -
New probe-level count matrix output files for Flex:
raw_probe_bc_matrix.h5andsample_raw_probe_bc_matrix.h5. -
The
frp_gem_barcode_overlap.csvprobe overlap file now contains content for Antibody Multiplexing Barcodes.
5’ Immune Profiling
-
New feature: Cell Ranger v7.2 supports the aggregation of BEAM (Antigen Capture) libraries with
cellranger aggrto combine and normalize the calculation of antigen specificity scores across multiple (or large samples) split across wells. -
The outputs of
cellranger aggrfor 5' Immune Profiling libraries now include theairr_rearrangement.tsv(not produced in previous versions of this pipeline). -
A bug that caused all alignments in the
consensus.bamandconcat_ref.bamfiles to have their POS field set to the default value of 1 has been fixed. -
The
all_contig_annotations.jsonoutput file has an additional field calledjunction_supportthat is a map of{reads: x, umis: y}supporting the junction region of a contig. This information is generated by thecellranger vdjassembler for productive contigs in reference-assisted assembly (or valid contigs in de novo assembly) and used for confidence determination and cell filtering.
General updates and bug fixes
-
Targeted Gene Expression analysis deprecated:
countandmultipipelines in v7.2 and later do not support the analysis of Targeted Gene Expression libraries. -
NovaSeq X demultiplexing support with bcl2fastq v2.20 dependency.
-
An new command line argument
--outputs-diris available forcount,multi,mkref,mkvdjref, andvdjpipelines to specify a custom output directory. -
Batch effect score calculations modified to normalize and scale with the number of cells in the dataset.
-
Cell Ranger now performs a preflight check to verify that the prebuilt reference file is complete. The use of an incomplete reference (due to incomplete download or corrupt file) produces an error message.
-
BAM tags update: MM changed to mm to enable compatibility with IGV.
-
BAM files now have @PG header noting which pipeline version was used to generate the file.
-
Improvements to the cellranger aggr web summary:
-
Layout improved.
-
Metrics table per library.
-
Provides aggregation summary for BEAM (Antigen Capture) libraries.
-
-
Improvements to the cellranger multi web summary:
-
In the Feature Barcode Expression Metrics table of the cells tab, the
Median UMI counts per cell metrichas been renamed in the Antibody tab:-
Antibody Capture:
Median antibody UMI counts per cell. -
CRISPR, Antigen Capture, and Custom library: remains unchanged.
-
-
Antibody histogram has moved from the library tab to the cells tab.
-
Flex new metrics to capture the fraction of reads mapping to two different halves of probes:
Reads half-mapped to probe set,Reads split-mapped to probe set.
-
-
Improvement to the Batch Effect Score (BES) calculation: normalization and scaling has changed from using √N nearest neighbors to 0.01*N nearest neighbors (with N being number of cells).
Changes that apply to Flex analysis
-
v1.0.1 probe set reference CSVs for human and mouse have a new region column, which indicates whether a probe spans a splice junction by at least 10 bp (spliced) or not (unspliced).
-
When the v1.0.1 probe set reference CSV is used in Cell Ranger v7.1, the web summary and
metrics_summary.csvfiles will include genomic DNA metrics. The region column information is used to calculate these metrics. -
The molecule_info.h5 files include the probe to which each molecule is mapped.
-
In the probe set reference CSV, the
includedcolumn is set toFALSEfor all deprecated probes.
General improvements
-
Calling cell barcode improvement: The auto-estimated
expect-cellsupper range has been restricted to the lower range of the EmptyDrops method. Previously, the upper range was 262,000 cells. The new upper range is 45,000 for single cell gene expression analyses. The exception is super loaded (sub-pooled) multiplex Flex analyses, where the range is calculated as:max(45,000, number of probe barcodes × 22,500). -
Improvement to the Batch Effect Score (BES) calculation to use √n nearest neighbors, where n is the total number of cells, instead of 100. Cells are no longer subsampled to 10%.
-
A new compression format,
.tar.xz, is available on the Cell Ranger downloads page. The smaller file size enables faster download. -
Cell Ranger 7.1 introduces a new subcommand,
cellranger multi-template, which provides descriptions for all multi config CSV parameters and produces a config CSV template. Runcellranger multi-template -hfor help. -
The
ARC-v1chemistry may be used to analyze only the Gene Expression library portion of a Multiome ATAC + Gene Expression experiment. -
The
aggregate_barcodes.csvoutput file for Antibody Capture analyses is no longer stored in aantibody_analysis/sub-directory. In the cellranger multi pipeline, it is found inouts/per_sample_outs/<sample_name>/count/aggregate_barcodes.csv. In thecellranger countpipeline, it is found in outs/aggregate_barcodes.csv. For both pipelines, the CSV file is only generated if antibody aggregates are detected. -
The web summaries for Antibody Capture libraries include a Distribution of Antibody Counts plot to show the relative composition of antibody counts for antibodies with at least one UMI.
Bug fixes
-
Fixed a bug in the OrdMag algorithm, which could result in all barcodes being called as cells when there are very few cells.
-
Improvement to 3' Cell Multiplexing tag assignment for samples with a large number of zero CMO UMI counts.
-
Fixed a 3' Cell Multiplexing t-SNE plot bug where the plots were generated assuming a full set of CMOs in
cmo-setinstead of those used in the experiment. -
Fixed conditions resulting in negative gap errors.
-
Fixed a bug that causes pipestance failure with an IOError message: "directory
%sexists but it can not be written".
The updates are explained further in these Knowledge Base articles:
- My samples are analyzed with Cell Ranger v7.0. Should I rerun analysis using the latest Cell Ranger v7.1?
- What are the cell calling updates in Cell Ranger v7.1 and its impact on Single Cell Gene Expression data?
- Should I upgrade Cell Ranger to v7.1 for cell multiplexing analysis?
New feature: Barcode Enabled Antigen Mapping (BEAM) or Antigen Capture
-
Cell Ranger 7.1 is required for the analysis of BEAM libraries. Instructions for running cellranger multi are described in the Antigen Capture page. The new
feature_type, antigen capture, the Feature Reference CSV that specifies the list of antigens (and MHC alleles) included in the experiment, and all the antigen specific customizable parameters in a multi config CSV are described in detail. Example multi config CSV for TCR and BCR Antigen Capture libraries are also provided. -
The algorithms section includes a page called Antigen Algorithms with a description of the new methods developed for processing Antigen Capture (BEAM) data.
-
If an Antigen Capture library is included, some new/updated output files are generated (described in the Understanding Outputs section):
antigen_specificity_scores.csv(new file)per_barcode.csv(new file)aggregate_barcodes.csv(updated location and format)
Changes that apply to 5' Immune Profiling analysis
-
V(D)J cell calling improvement: If a Gene Expression library is present, the V(D)J cell calling algorithm does not filter out two or more clonotypes that have identical chains. This helps improve V(D)J cell calling, especially for transgenic strains. This change does not apply to V(D)J datasets in the absence of a Gene Expression library.
-
The Human V(D)J reference has been updated to exclude the following genes:
- IGHV4-30-2
- IGKV1D-33
- IGKV1D-37
- IGKV1D-39
- IGKV2D-28
These genes have counterparts with identical V, D, J, and C gene sequences, but differ in the length of their 5' UTRs. Removing duplicates improves clonotype assignment.
Updates are explained further in this Knowledge Base article: What are the major updates in Cell Ranger v7.1 that impacts V(D)J data?
Bug fixes
-
Fixed a bug where an upgrade to Illumina NovaSeq control software v1.8 (reagent name change in recipe XML file) resulted in a silent
cellranger mkfastqerror and a significant number of reads going intoUndetermined/because the orientation of i5 (Index2) could not be autodetected. -
Improved Deplex Error message for
cellranger multiwhen no valid cell multiplexing tags are detected in the Multiplexing Capture library. Common failure modes are provided to help with troubleshooting. -
Updated Flex web summary metric names and definitions for consistency.
New feature: Flex
- Cell Ranger 7.0 is required for analysis of Flex data. Instructions for running the
cellranger multisubcommand are described in the running multi pipeline page. This includes a new option,probe-set, to specify the probe set CSV file. Output files are described in the Understanding Outputs section. The Flex algorithms section includes descriptions of the new methods that were developed for processing Flex data.
Major updates
-
To maximize sensitivity for whole transcriptome 3’/5’ Single Cell Gene Expression and 3’ Cell Multiplexing experiments, introns will be included in the analysis by default for
cellranger countandmulti. There will be an informational alert in the count and multi web summaries to indicate that intronic reads were included in your analysis. While not recommended, users can exclude introns by settinginclude-introns=falsein Cell Ranger. This change does not apply to the 3’/5’ Targeted Gene Expression or Flex assays, as both target exonic sequences. Learn more. -
CRISPR Guide Capture libraries can be aggregated with
cellranger aggr. This addition allows users to combine large CRISPR assays across multiple GEM wells. There are no changes toaggrinputs – the presence of CRISPR libraries in themolecule_info.h5input files enables CRISPR aggregation. Normalization is enabled by default for both Gene Expression and CRISPR libraries; changes to the normalization parameters affect both libraries. Protospacer calling is performed again on the combined data included in thecellranger aggrrun. CRISPR aggregation generates thecrispr_analysis/folder in theouts/directory. The structure of the crispr_analysis folder is similar to the CRISPR outputs fromcount.
General improvements
-
Users no longer need to specify
expect-cellsforcellranger countandmultipipelines due to improvements in the gene expression cell calling algorithm. The expected number of cells can either be auto-estimated (recommended) or users can still provide a reasonable estimate toexpect-cells. -
The new
check-library-compatibilityoption allows users to disable the default check for 10x Barcode overlap when multiple libraries are specified for cellranger count and multi (3' Gene Expression, 5' Immune Profiling). -
For 3’ Cell Multiplexing analysis in
cellranger multi, users can override Cell Ranger’s cell calling and tag calling algorithms with the custom cell assignment input file specified by thebarcode-sample-assignmentoption in the multi config CSV file. -
Modifications to the 3’ Cell Multiplexing CMO tag calling algorithm enable users to recover viable singlet data from “blank” assignments.
-
The following per-sample output files from
cellranger multihave been renamed:
| Cell Ranger 6.1.2 outputs | Cell Ranger 7.0 outputs |
|---|---|
cloupe.cloupe | sample_cloupe.cloupe |
sample_barcodes.csv | sample_filtered_barcodes.csv |
sample_feature_bc_matrix | sample_filtered_feature_bc_matrix |
sample_feature_bc_matrix.h5 | sample_filtered_feature_bc_matrix.h5 |
- Secondary analysis outputs will be named to reflect which library they are specific to (
gene_expression_*,antibody_capture_*,crispr_guide_capture_*,multiplexing_capture_*). The secondary analysis clustering, PCA/t-SNE/UMAP, and differential gene expression outputs are supported for Gene Expression and Antibody Capture libraries, while PCA/t-SNE/UMAP outputs are supported for CRISPR Guide Capture and Cell Multiplexing libraries. For example:
└── analysis
└── pca
├── antibody_capture_10_components/
└── gene_expression_10_components/
-
The
cellranger countweb summary “Analysis” tab has been renamed to “Gene Expression”. There is an “Antibody” tab for Antibody Capture analysis, which includes a t-SNE projection plot by clustering and a histogram of antibody counts. -
The
cellranger multiweb summary (3' Gene Expression, 5' Immune Profiling) “Sample” view has been renamed to “Cells”. The “Antibody” tab includes a t-SNE projection plot by clustering. The mapping metrics, sequencing saturation plot, and median genes per cell plot are displayed on the “Library” view (previously appeared on “Sample” and “Library” view). -
Cell Ranger can now ingest FASTQs with a quality score up to the full supported range (93 instead of 41).
Bug fixes
-
Improved error messages and better handling of poorly formatted inputs in
cellranger mkref. Enable users to generate references for analyses with large genomes containing chromosomes longer than 512 Mbp.cellranger countandmultipipelines will output a.csiBAM index file instead of .bai in these cases. -
Fixed a bug that resulted in a segmentation fault error when mapping to references that contain small contigs, for example, the rabbit genome.
-
Removed the
Inconsistent Throughput Detectedalert in web summary when it should not appear. -
Fixed a bug where
vdjpipeline failed for specific CentOS/RHEL 7 kernels. -
Bundles the latest version of
bamtofastq(v1.4.1) in Cell Ranger 7.0 tarball. -
Fixed a bug where
bamtofastqfailed if the R1 read length was >26bp.
Changes that apply to 5' Immune Profiling analysis
-
Support for gamma-delta libraries: The
cellranger multipipeline can process T cell receptor (TCR) libraries enriched for gamma (TRG) and delta (TRD) chains. 10x Genomics does not officially support TRG/D analysis with a reagent kit. Please note that, only CDR3 annotation is available for TRG/D, and the quality of annotations cannot be guaranteed. Users must specifyVDJ-T-GDas thefeature_typein thecellranger multiconfig CSV as TRG/D chains cannot be autodetected. Aweb_summaryalert is displayed to indicate the use of an unsupported workflow. No TRG/D analysis is available via thecellranger vdjpipeline. -
V(D)J Reference updated: The recommended V(D)J reference packages for human and mouse have been updated from version 5.0 to 7.0. The changes to the V(D)J reference sequences are listed below:
HUMAN:
-
Added human IGHV3-9
-
For two genes that are identical except for extra bases on the 3' end, only the longer version was retained. List of affected genes:
IGHA1 ENST00000390547 IGHD ENST00000390556 IGHD ENST00000390556 IGHD1-1
ENST00000454908 IGHD1-14 ENST00000451044 IGHD1-20 ENST00000450276 IGHD1-26
ENST00000390567 IGHD1-7 ENST00000430425 IGHD1/OR15-1A ENST00000605284 IGHD2-15
ENST00000390578 IGHD2-2 ENST00000390591 IGHD2-21 ENST00000390572 IGHD2-8
ENST00000390585 IGHD2/OR15-2A ENST00000603077 IGHD3-10 ENST00000390583
IGHD3-16 ENST00000390577 IGHD3-22 ENST00000390571 IGHD3-3 ENST00000390590
IGHD3-9 ENST00000390584 IGHD3/OR15-3A ENST00000604950 IGHD4-11 ENST00000431440
IGHD4-17 ENST00000431870 IGHD4-23 ENST00000437320 IGHD4/OR15-4A
ENST00000603326 IGHD5-12 ENST00000390581 IGHD5-18 ENST00000390575 IGHD5-24
ENST00000390569 IGHD5/OR15-5A ENST00000604642 IGHD6-13 ENST00000390580
IGHD6-19 ENST00000390574 IGHD6-25 ENST00000452198 IGHD6-6 ENST00000454691
IGHD7-27 ENST00000439842 IGHG1 ENST00000390542 IGHG1 ENST00000390548 IGHG1
ENST00000390549 IGHG2 ENST00000390545 IGHG3 ENST00000390551 IGHG4
ENST00000390543 IGHJ1 ENST00000390565 IGHM ENST00000390559 IGHV1-18
ENST00000390605 IGHV1-2 ENST00000390594 IGHV1-24 ENST00000390610 IGHV1-3
ENST00000390595 IGHV1-45 ENST00000390621 IGHV1-46 ENST00000390622 IGHV1-58
ENST00000390628 IGHV1-69 ENST00000390633 IGHV1-69-2 ENST00000615784 IGHV2-26
ENST00000390611 IGHV2-5 ENST00000390597 IGHV2-70D ENST00000390634 IGHV3-11
ENST00000390601 IGHV3-13 ENST00000390602 IGHV3-15 ENST00000390603 IGHV3-16
ENST00000390604 IGHV3-20 ENST00000390606 IGHV3-21 ENST00000390607 IGHV3-23
ENST00000390609 IGHV3-30 ENST00000603660 IGHV3-35 ENST00000390617 IGHV3-38
ENST00000390618 IGHV3-43 ENST00000434710 IGHV3-48 ENST00000390624 IGHV3-49
ENST00000390625 IGHV3-53 ENST00000390627 IGHV3-64 ENST00000454421 IGHV3-66
ENST00000390632 IGHV3-7 ENST00000390598 IGHV3-72 ENST00000433072 IGHV3-73
ENST00000390636 IGHV3-74 ENST00000424969 IGHV4-28 ENST00000390612 IGHV4-34
ENST00000390616 IGHV4-39 ENST00000390619 IGHV4-4 ENST00000455737 IGHV4-59
ENST00000390629 IGHV4-61 ENST00000390630 IGHV5-51 ENST00000390626 IGHV6-1
ENST00000390593 IGKV1-12 ENST00000480492 IGKV1-16 ENST00000479981 IGKV1-17
ENST00000490686 IGKV1-27 ENST00000498435 IGKV1-33 ENST00000473726 IGKV1-37
ENST00000465170 IGKV1-39 ENST00000498574 IGKV1-5 ENST00000496168 IGKV1-6
ENST00000464162 IGKV1-8 ENST00000495489 IGKV1-9 ENST00000493819 IGKV2-24
ENST00000484817 IGKV2-28 ENST00000482769 IGKV2-30 ENST00000468494 IGKV3-11
ENST00000483158 IGKV3-15 ENST00000390252 IGKV3-20 ENST00000492167 IGKV3-7
ENST00000390247 IGKV3D-7 ENST00000443397 IGKV5-2 ENST00000390244 IGKV6-21
ENST00000390256 IGLV1-36 ENST00000390301 IGLV1-40 ENST00000390299 IGLV1-44
ENST00000628287 IGLV2-33 ENST00000390302 IGLV3-32 ENST00000390303 IGLV5-37
ENST00000390300 IGLV7-43 ENST00000390298 TRBD1 ENST00000631435 TRBJ1-1
ENST00000634213 TRBJ1-2 ENST00000631745 TRBJ1-3 ENST00000633780 TRBJ1-4
ENST00000632041 TRBJ1-5 ENST00000634000 TRBJ2-1 ENST00000390412 TRBJ2-2
ENST00000390413 TRBJ2-2P ENST00000390414 TRBJ2-3 ENST00000390415 TRBJ2-4
ENST00000390416 TRBJ2-5 ENST00000390417 TRBJ2-6 ENST00000390418 TRBV10-1
ENST00000390364 TRBV11-1 ENST00000390367 TRBV11-3 ENST00000611787 TRBV12-3
ENST00000620569 TRBV13 ENST00000614171 TRBV14 ENST00000617639 TRBV15
ENST00000616518 TRBV16 ENST00000620773 TRBV23-1 ENST00000390396 TRBV27
ENST00000390399 TRBV28 ENST00000390400 TRBV29-1 ENST00000422143 TRBV3-1
ENST00000390387 TRBV4-2 ENST00000390392 TRBV5-1 ENST00000390381 TRBV5-6
ENST00000390375 TRBV5-7 ENST00000390378 TRBV6-1 ENST00000390353 TRBV6-5
ENST00000390368 TRBV7-1 ENST00000547918 TRBV7-7 ENST00000390377 TRGJ1
ENST00000390337
MOUSE
- Added missing mouse TRGV and TRGC genes
TRGC1 ENSMUST00000103558 TRGC2 ENSMUST00000103561 TRGC3 ENSMUST00000198163
TRGC4 ENSMUST00000179181 TRGV1 ENSMUST00000103564 TRGV3 ENSMUST00000198663
TRGV4 ENSMUST00000103554 TRGV5 ENSMUST00000199017 TRGV6 ENSMUST00000198330
TRGV7 ENSMUST00000103553
- For two genes that are identical except for extra bases on the 3' end, only the longer version was retained. List of affected genes:
IGHD2-5 ENSMUST00000178549 IGHD5-2 ENSMUST00000179166 TRAV11D
ENSMUST00000103648 TRAV12D-1 ENSMUST00000181360 TRAV12D-2 ENSMUST00000197007
TRAV13D-2 ENSMUST00000197954 TRAV14D-1 ENSMUST00000181038 TRAV14D-2
ENSMUST00000196802 TRAV15D-2-DV6D-2 ENSMUST00000199800 TRAV3D-3
ENSMUST00000196023 TRAV4D-3 ENSMUST00000103592 TRAV4D-4 ENSMUST00000103600
TRAV5D-4 ENSMUST00000179701 TRAV6-6 ENSMUST00000103584 TRAV7-2
ENSMUST00000103636 TRAV7D-5 ENSMUST00000197128 TRAV9D-2 ENSMUST00000199746
TRAV9D-3 ENSMUST00000178252
-
V(D)J web summary: In the
web_summary.htmlfile produced bycellranger vdj, the Analysis tab has been renamed to VDJ. -
The default for the
fiveprime_multiplexingparameter in thecellranger-7.0.0/lib/bin/parameters.tomlfile has changed toTrue.
Bug fixes
-
Improved handling of memory requests for large genomes.
-
Fixes an issue in the
molecule_info.h5file, where if a species was not present in a barnyard run, it was omitted from the genome information. -
Reduces the size of the bundled reference included in
cellranger testrun. -
Adds additional guidance in the web summary when there is a low fraction of targeted genes enriched.
-
Fixes a metric issue where aggregate antibodies could be double-counted.
-
Unsets additional sysconfig environment variables prior to pipeline execution, which may otherwise interfere with the pipeline conda environment.
Bug Fixes
- Fix an issue where
cellranger vdjcould fail if executed by a user without a home directory.
New Feature: High Throughput (HT) for Chromium X
-
Cell Ranger 6.1 introduces support for the 3' and 5' High Throughput (HT) kits with 16 channels per chip, allowing users to process 2,000-20,000 cells per channel (3' and 5') or 2,000-60,000 cells per channel with CellPlex (3' only). HT kits are only compatible with Chromium X, which is backwards compatible with all 10x Genomics dual indexed assays. For more information see What is HT?
-
Cell Ranger 6.1 includes a new throughput detection algorithm to detect HT samples in 3' CellPlex data as described in the CellPlex algorithms page. In the event that chemistry detection fails, it can be overridden with the option (e.g.,
--chemistry=SC3Pv3HT) incellranger count, or in thecellranger multiCSV file to detect HT samples when 3' CellPlex libraries are run. -
Minor changes to web_summary.html include a new alert when the user specifies
--chemistry=SC3Pv3HT, but Cell Ranger detects otherwise. HT will be appended to the detected chemistry if the pipestance was a multiplexing run. -
Note that with HT chemistry and 3' CellPlex it is now possible to run 60,000 cells per GEM well, and aggregate million-cell datasets. Larger datasets will require additional memory beyond our stated minimum requirement of 64 GB. See the 3' system requirements page for more details and time trial data.
General Improvements
-
Numerous performance optimizations have been made, especially for pipeline stages that iterated over
molecule_info.h5files, such ascellranger aggr, but also memory allocation improvements. We have seen up to 2-3x speed improvements forcellranger aggr. -
Changed certain parameters in the cell calling algorithm for improved results. Changed the Empty Drops stage for multispecies experiments to call cells using only the UMI counts for each species separately.
-
Raw feature barcode matrices are no longer output by
cellranger aggr. It is no longer possible to specify--force-cellsof an aggr output incellranger reanalyzewith more cells than were originally called. -
The secondary analysis implementation is now shared with Loupe Browser's Filtering and Reclustering Wizard. These changes improve the performance of most stages, in either time (t-SNE) or memory (PCA). There will be changes in outputs compared to previous versions, reflecting either slight variations in outputs (PCA, t-SNE), or as if a different randomized seed had been chosen (graph clustering, UMAP).
-
Starting from Cell Ranger 6.1, antibody histograms of UMI counts are shown on Library tab of the
web_summary.html, and protein aggregate barcodes are provided asaggregate_barcodes.csv. These are meant to help feature barcoding users diagnose issues of aggregating antibodies on cell surface proteins. -
Fixed a bug that caused Cell Ranger v4 and higher to ignore user-supplied parameters to
--nthreads, defaulting to 1. Parallelization has been re-enabled in Cell Ranger 6.1.
Deprecating OS
- The recommended operating systems for Cell Ranger v6.1 are CentOS 7 or Ubuntu 14 Linux variants or newer. CentOS 6 and Ubuntu 12 are still supported but have been deprecated (unsupported for future Cell Ranger releases). Support may be dropped in future versions.
Bug fixes
-
Fixes additional issues with file copying on BeeGFS filesystems.
-
cellranger multi: Adds an optionalmin-assignment-confidencein the config CSV to allow adjustment of the Cell Multiplexing minimum assignment confidence threshold (default: 0.9). Decreasing the threshold will likely increase the number of singlets assigned to samples, but at the cost of potentially increasing the rate of mis-assignment. -
Adds a warning to the
cellranger multiweb_summary.htmlif contaminant tags are detected in Cell Multiplexing experiments.
General improvements
-
The fetch-imgt script, to build an IMGT-compatible custom reference for Single Cell Immune Profiling data analysis, has been updated to be compatible with Python 3.
-
Cell Multiplexing analysis has been updated to be more memory-efficient.
Bug fixes
-
The [sample] section of the configuration CSV file is now required for Cell Multiplexing analysis.
-
Fixes an issue where
cellranger multiwould only accept a single VDJ library. -
Fixes an issue where
cellranger vdjpreflight would fail if custom primers were passed in. -
The
sample_idinformation in Cell Ranger 6 aggr runs are now correctly propagated to Loupe Browser. -
Fixes an issue where
.vloupefiles fail to generate on some filesystems and operating systems. -
Fixes an issue in cluster mode where the pipeline could fail to correctly identify which jobs were still queued.
-
Fixes an issue where including an
aggrcsv inreanalyzewould cause the pipeline to exit. -
The "Number of reads for Custom Feature by Physical library ID" in the
multiweb summary and metrics summary is now rendered properly. -
Fixes an issue with file copying on BeeGFS filesystems.
New Feature: Cell Multiplexing
-
Cell Ranger 6.0 now supports analysis of Cell Multiplexing data for the 3' Gene Expression, Targeted Gene Expression, and Feature Barcode solutions. Instructions for running the
cellranger multisubcommand are described in the running multi page. A new Getting Started Tutorial is also available. The Cell Multiplexing algorithms include a new method to call singlets, multiplets, and empty drops. The output file structure has also changed to accommodate multiple samples multiplexed in a single GEM well. -
The aggr subcommand now supports analysis of
cellranger multioutputs for the 3' Gene Expression, Targeted Gene Expression, and Feature Barcode solutions. Further details are described in the running aggr page.
New Feature: LT (Low Throughput) support
- Cell Ranger 6.0 supports the analysis of data from 3' Gene Expression and Feature Barcode (Cell Surface Protein) LT (Low Throughput) kits.
Changes that apply to Gene Expression and Feature Barcode analysis
-
The column names for the
Aggregation CSVfile required by theaggrsub-command have changed:library_idhas been changed tosample_idandlibrary_outshas been changed tosample_outs. Further details are described in the running aggr page. -
The
molecule_info.h5and unfiltered feature-barcode matrix files (raw_feature_bc_matrixin H5 and MEX formats) will only contain barcodes with at least one read, rather than all barcodes in the inclusion list. -
The change to the unfiltered feature-barcode matrix summarized in (4) above results in a subtle change to the distribution of UMI counts amongst background, i.e. non-cell barcodes, which results in minor changes to the results of the cell calling algorithm. This change occurs due to the second step that identifies non-ambient cell-barcodes as described in the algorithms page.
-
Cell Ranger 6.0 is the first Cell Ranger release to use Python 3.
Bug fixes and deprecations
-
A bug has been fixed in the graph-based clustering output: previously, in a sample with K clusters, the first K cell-associated barcodes (ordered as in the filtered feature-barcode matrix) may have been assigned incorrect cluster labels. This change does not affect the number of clusters output.
-
A bug has been fixed for multi-genome experiments, wherein the species annotation may have been incorrect for cell-associated barcodes identified by the second step of the cell-calling algorithm, as described in the algorithms page. Changes in metrics are expected to be minor, unless the the proportion of such cells is large.
-
The
--qcoption has been deprecated fromcellranger mkfastq. -
A bug has been fixed for multi-genome experiments, wherein the species annotation may have been incorrect for cell-associated barcodes identified by the second step of the cell-calling algorithm, as described in the algorithms page. Changes in metrics are expected to be minor, unless the the proportion of such cells is large.
Changes that apply to 5' Immune Profiling analysis
In Cell Ranger 6.0, the following changes apply to joint analysis of Immune Profiling, Gene Expression, and Feature Barcode data with the multi sub-command:
-
The structure of the
outs/folder has been updated, as described in runningcellranger multi. -
When running the
cellranger aggrsubcommand on samples that have Immune Profiling, Gene Expression, and/or Feature Barcode data analyzed with multi, thesample_outsfield now contains the path to the outputs for that sample (e.g.outs/per_sample_outs/sample_x). Further details are described in running aggr.
Cell Ranger 6.0 also introduces some improvements and bug fixes related to the clonotype inference algorithms:
-
There are subtle changes to clonotyping heuristics that have little effect on overall behavior, but recover a small number of joins that were previously missed and might be critical for a particular experiment. These changes are described in terms of technical parameters to the algorithm, specifically raising the default for
MAX_DIFFSfrom 50 to 55 and raising the default forMAX_CDR3_DIFFSfrom 10 to 15. There were also compensatory changes to prevent the rate of false positive joins from increasing: the default forMAX_DEGRADATIONwas lowered from 3 to 2, and the default forMAX_SCOREwas lowered from 1,000,000 to 500,000. For more details, visit enclone help. -
Single-chain clonotypes are now more likely to be merged with two-chain and three-chain clonotypes. This causes significantly more clonotypes to have single-chain exact subclonotypes.
-
Fixed a bug that caused failures on some very short (defective) V gene reference sequences.
-
The algorithm for deciding to use a donor reference allele now checks all donor reference alleles for all V genes having the same name as the one originally assigned to a contig. For more details, visit enclone help.
-
A doublet test has been added. This removes some exact subclonotypes that appear to represent doublets. Details are documented on the enclone pages. The typical effect is to remove some three-chain and four-chain clonotypes, with the fraction removed depending on the emperical doublet rate. In some cases, large, complex clonotypes are accurately split into multiple smaller clonotypes by this change.
-
There is no longer a restriction on the length of CDR3 sequences (previously maximum 27).
-
The Immune Profiling output file
all_contig_annotations.csvcontains new fieldsfwr1, ..., fwr4andcdr1,cdr2, providing the amino acid sequences of framework and complementarity-determining regions (in addition tocdr3, which was already present). The definitions used to define these regions are provided in the enclone features page. The corresponding nucleotide sequences are provided (e.g.fwr1_nt). These fields are also provided in the fileconsensus_annotations.csv, as are nucleotide start and end positions (e.g.fwr1_start). -
The Immune Profiling output file
all_contig_annotations.csvcontains new fieldexact_subclonotype_idproviding the exact subclonotype ID to which the cell barcode was assigned. Details about exact subclonotypes can be found on the clonotype grouping page. -
The
--qcoption has been deprecated fromcellranger mkfastq.
Bug fixes
-
Fixes an issue in aggr where files would fail to be copied on NFSv4 File Systems.
-
Fixes an issue in multi where r1-length and r2-length settings were ignored for
vdj.
Changes that apply to Gene Expression and Feature Barcode analysis
-
Cell Ranger v5.0 introduces a
--no-bamoption that disables the generation of aligned BAMs for gene expression and feature barcode datasets. If you have no need for these files, then disabling their generation can significantly speed up the pipeline. -
Cell Ranger v5.0 introduces upgraded protein aggregation detection and filtering algorithm. By directly using the protein counts, more aggregate GEMs are detected and filtered out before proceeding with cell calling.
-
Cell Ranger v5.0 introduces an
--include-intronsoption for counting intronic reads using 3’ Gene Expression and 5’ Gene Expression products. The usage of pre-mRNA references for counting intronic reads is now deprecated.- The
--include-intronsoption, introduced in Cell Ranger 5.0, works by aligning reads to a normal reference transcriptome with STAR. After alignment, the reads mapping to introns are annotated and counted similarly to reads that are aligned to exons. Previously, the pre-mRNA reference strategy implemented with Cell Ranger 4.0 and earlier involves alignment to a modified reference transcriptome that categorizes intronic regions as exonic. There are slight differences in read alignments produced by the STAR aligner when a pre-mRNA reference is used compared to a normal reference using--include-introns. These differences result in small overall differences in counted UMIs for intron-mode compared to pre-mRNA-reference.
- The
-
Ported a fix from upstream
IRLBAthat fixes incorrect behavior in rare circumstances. -
On some Linux distributions, NFS implementations would surface an improper error during file copy. We have implemented a workaround for our affected native code.
Changes that apply to Gene Expression, Feature Barcode, and V(D)J analysis
-
Cell Ranger 5.0 introduces the multi pipeline that can simultaneously process any combination of 5' Gene Expression, Feature Barcode (cell surface protein or antigen) and V(D)J libraries from a single GEM well. The multi pipeline uses the cell calls provided by the gene expression data to improve the cell calls inferred by the V(D)J library.
-
A new metric, “Number of Short Reads Skipped”, is added to the web summary, indicating the total number of read pairs that were ignored by the pipeline because they do not satisfy the minimum length requirements.
Changes that apply to V(D)J analysis
-
Cell Ranger v5.0 introduces a new clonotype grouping algorithm that computationally approximates groups of cells which are descendants of a single, fully rearranged common ancestor and infers the germline sequence of the V genes from each individual in the dataset. In previous versions (4.0 and earlier), the algorithm grouped cells based only on the set of productive CDR3 nucleotide sequences. As a consequence, whenever a true clonotype had a CDR3 mutation, the true exact subclonotypes were presented by the algorithm as multiple separate clonotypes. The previous approach to clonotyping in Cell Ranger 4.0 and earlier led to inaccuracies in the B cell clonotypes due to the grouping by unique CDR3 sequence. Additionally, single-chain clonotypes were reported as separate clonotypes, which could lead to both over- and under-estimation of the size of a given clonotype. The new clonotyping algorithm is improved in specificity, sensitivity, and overall accuracy because it accounts for mutations found in the V(D)J transcript and in the V(D)J junction. It also merges single chain clonotypes into the correct fully-paired clonotypes for both T cells and B cells. Additional cell filters are also imposed during clonotyping to improve data quality.
-
Changes to V(D)J outputs:
-
The following output files are removed in 5.0:
consensus.fastqandconsensus_annotations.json -
The following output files are added in 5.0: - Contig info binary file, which would be used as an input to aggregate V(D)J samples - Donor reference fasta
-
Two new columns are added to the clonotypes.csv file that displays the iNKT/MAIT evidence.
-
The files
filtered_contig_annotations.csv,filtered_contig.fasta,filtered_contig.fastqnow only contain data from the contigs in cell barcodes that are productive. -
A number of new fields are added to
consensus_annotations.csv:v_start,v_end,v_end_ref,j_start,j_start_ref,j_end,cdr3_start,cdr3_end
-
-
The recommended V(D)J reference packages for human and mouse have been updated from v4.0-5.0. The changes to the V(D)J reference sequences are listed below:
HUMAN:
- Replace IGKV2D-40, whose leader sequence appears to be truncated.
- Delete IGKV2-18, which is probably a pseudogene.
- Delete IGLV5-48, which is truncated on the right.
- Delete TRBV21-1, which has multiple frameshifts.
- Add IGHV4-30-4, which was missing.
- Add IGKV1-NL1, which was missing.
- Add IGHV4-38-2, which was missing.
MOUSE:
- Delete TRAV23, which is frame-shifted.
- Delete the first base of the constant region gene IGHG2B.
- Make a six-base insertion in IGKV12-89, based on empirical data.
- Correct IGHV8-9, whose amino acid sequence showed the canonical C at the end of FWR3 as S. This is consistent with 10x data.
- Add an allele of IGKV2-109, which was missing.
- Add IGKV4-56, which was missing.
- Add IGHV1-2, which was missing.
-
cellranger aggrnow aggregates V(D)J data, allowing users to recompute V(D)J clonotype groupings across the combined data. -
Soft deprecation of
--force-cellsincellranger vdj:-
Since Cell Ranger 3.1, due to filters in the VDJ assembler,
--force-cellsin VDJ pipelines did not behave as users would expect it to behave. Users can only apply--force-cellsto the number of barcodes passing the combined filters in the assembler. -
This makes it effectively impossible for users to increase the number of recovered cells. Rather, it is only possible to reduce the number of recovered cells using
--force-cellsin this context, unlike the behavior in thecellranger countpipeline. -
Because this specific flag is likely to be misunderstood by users, and is also not highly requested, we are starting to deprecate it. In Cell Ranger 5.0,
--force-cellswill be available only as an undocumented silent option. This will also allow users who are using this routinely in their workflows to anticipate eventual deprecation.
-
Changes that apply to Gene Expression and Feature Barcode analysis
-
Targeted Gene Expression analysis is available in Cell Ranger 4.0 and is invoked by specifying the
--target-paneloption when running the cellranger count command. -
Cell Ranger 4.0 introduces the new
targeted-comparepipeline for direct comparative analysis of matched parent Whole Transcriptome Amplification (WTA) and Targeted Gene Expression datasets. -
Cell Ranger 4.0 includes the new
targeted-depthsubcommand to estimate sequencing depths appropriate for Targeted Gene Expression experiments based on input WTA results and an associated target panel file. -
Recommended reference packages for human and mouse have been updated from version 3.0.0 to 2020-A:
-
Transcriptome annotations updated from Ensembl 93 to GENCODE v32 (human) and vM23 (mouse), which are equivalent to Ensembl 98.
-
GRCh38 and mm10 sequences are not changed; chromosome names now follow the GENCODE/UCSC convention (e.g.,
chr1andchrM) rather than the Ensembl convention (1andMT). -
Additional filtering removes genes with unreliable annotations that often overlap more legitimate genes (see build scripts for details), resulting in improved overall sensitivity. 2020-A reference packages are backwards compatible with Cell Ranger v3.1.0 and prior.
-
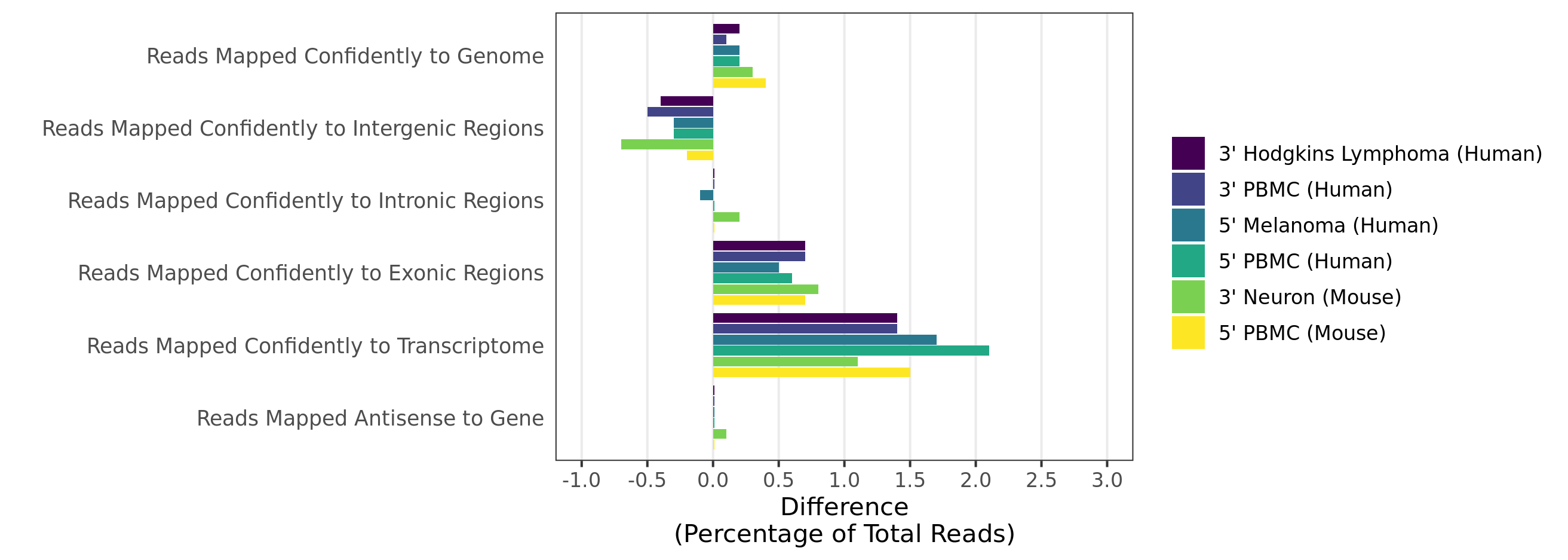
Mapping rates and gene/UMI sensitivity are increased due to more comprehensive annotations and improved manual curation of genes:
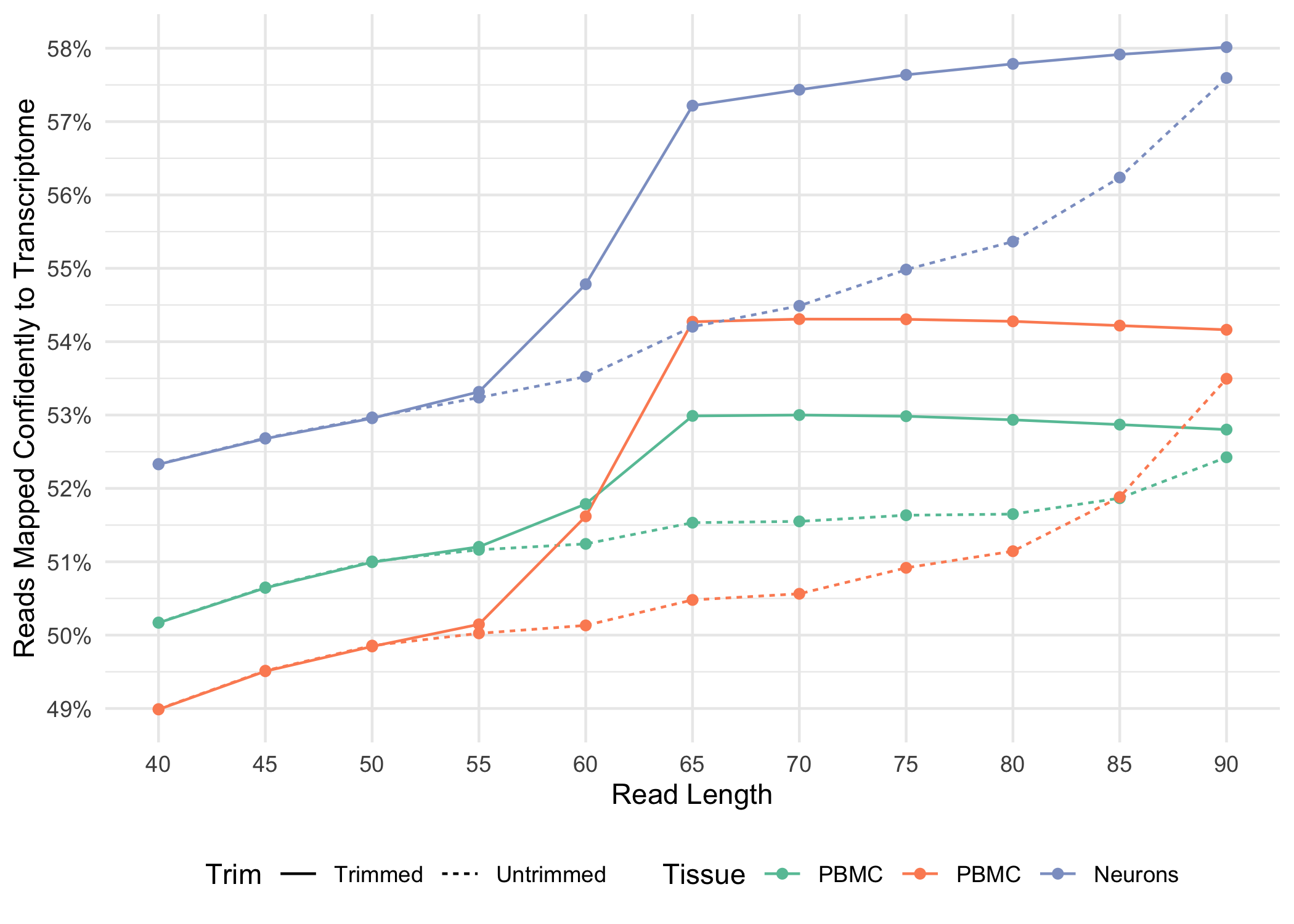
- When analyzing 3’ Gene Expression data, Cell Ranger 4.0 trims the template switch oligo (TSO) sequence from the 5’ end of Read-2 and the poly-A sequence from the 3’ end before aligning reads to the reference transcriptome. This behavior is different from Cell Ranger 3.1, which does not perform any trimming.
A full length cDNA molecule is normally flanked by the 30-bp TSO sequence, AAGCAGTGGTATCAACGCAGAGTACATGGG, at the 5' end and the poly-A sequence at the 3' end. Some fraction of sequencing reads are expected to contain either or both of these sequences, depending on the fragment size distribution of the library. Reads derived from short RNA molecules are more likely to contain either or both TSO and poly-A sequence than longer RNA molecules.
Trimming results in better alignment, with the fraction of reads mapped to a gene increasing by up to 1.5%, because the presence of non-template sequence in the form of either TSO or poly-A confounds read mapping. Trimming improves the sensitivity of the assay as well as the computational efficiency of the pipeline. Tags ts:i and pa:i in the output BAM files indicate the number of TSO nucleotides trimmed from the 5' end of Read-2 and the number of poly-A nucleotides trimmed from the 3' end. The trimmed bases are present in the sequence of the BAM record and are soft clipped in the CIGAR string.
Below, we illustrate how the fraction of reads mapped confidently to the transcriptome varies for both trimmed and untrimmed alignment as a function of read-length for a variety of sample types .
-
Cell Ranger 4.0 adds support for an “un-tethered” Feature Barcode pattern, (BC) without an anchor, specified in the Feature Reference CSV. This option allows the user to specify the sequence of the Feature Barcode without specifying a particular location on the read where the sequence is expected to be found.
-
cellranger reanalyzenow outputs the count matrix used in the analysis, so as to reflect any subsetting of barcodes used. -
Bug fixes for GTF files output by
mkref. These changes do not affect the pipeline results.- GTF attributes with duplicate keys (e.g., tag
"value1";tag "value2";) are handled correctly. Previously, only the last such attribute was kept. - GTF attributes with unquoted integer values (e.g.,
exon_number 1;) are kept. Previously, they were removed. - GTF lines end with semicolons.
- Unix line endings are used rather than DOS line endings, consistent with other Cell Ranger outputs.
- GTF attributes with duplicate keys (e.g., tag
-
Bug fixes for the BAM file
- The duplicate flag (
0x400) is set correctly in the secondary alignments (flag0x100) of PCR duplicate reads and low-support UMI reads (xf:i:2) - Low-support UMI reads (
xf:i:2) have the corrected barcode in UB:Z. Previously, it contained the raw barcode.
- The duplicate flag (
-
BAM file changes
- Cell Ranger v4.0 will not output the
li:itag. TheRG:Ztag contains this information. - Cell Ranger v4.0 will not output the
BC:ZandQT:Ztags.
- Cell Ranger v4.0 will not output the
-
Cell Ranger v4.0 now relies on Orbit to perform transcriptome alignment, which leverages a modified STAR v2.7.2a. These modifications provide compatibility with “versionGenome 20201” references, such as those generated by STAR v2.5.1b. In Cell Ranger 4.0 we still provide and use STAR v2.5.1b for other purposes such as
cellranger mkref. In our testing we did not note any differences in transcriptome alignments between the STAR shipped in Cell Ranger 3.1 (STAR v2.5.1b), STAR v2.7.2a, or Orbit. -
mkfastqnow accepts file names without lane number, e.g.,sample1_S1_R1_001.fastq.gz. -
Cell Ranger's
aggrpipeline no longer supports the aggregation of v1mol_info.h5files.
Changes that apply to Gene Expression, Feature Barcode, and V(D)J analysis
-
mkfastqsupports dual-indexed libraries for gene expression, both WTA and Targeted, V(D)J, and Feature Barcode datasets. -
mkfastqsupports a new sequencing configuration for Novaseq where the I2 index may need to be reverse-complemented before demultiplexing dual-indexed libraries. -
mkfastqnow accepts file names without lane number, e.g., sample1_S1_R1_001.fastq.gz. -
countandvdjrun approximately two to four times faster than in Cell Ranger v3.1, depending on the sequencing data, and reduces disk I/O by half. -
A new command-line interface with improved error-handling has been engineered into Cell Ranger v4.0.
-
The Martian pipeline framework has been upgraded to v4.0.
mrpandmrjobwill shut down if they detect that their log files were deleted or renamed. See the Martian release notes for more details. -
The following features present in Cell Ranger v3.1 are no longer present in Cell Ranger 4.0:
mkfastqno longer supports data from the Single Cell 3′ v1 chemistry.- The
cellranger demuxsubcommand has been removed. - The command-line interface does not accept FASTQs created by the deprecated cellranger demux pipeline. If you need to process FASTQs in this layout, contact support@10xgenomics.com for assistance.
cellranger countandcellranger vdjare no longer able to process data from multiple gem-wells through manual editing of MRO files. The Single Cell 3′ v1 and Single Cell 5′-R1 assay configurations will no longer be autodetected in Cell Ranger 4.0. Users who want to analyze data from those chemistries must explicitly specify the chemistry (SC3Pv1orSC5P-R1respectively) using the--chemistryargument.- The
--idargument used by the pipelines has a 64 character limit in Cell Ranger 4.0.
-
The
--idargument used by the pipelines has a 64 character limit in Cell Ranger 4.0.
Changes that apply to V(D)J analysis
-
Recommended VDJ reference packages for human and mouse have been updated from version 3.1.0 to 4.0.0. The changes to the VDJ reference sequences are listed below:
- Remove the first base of the C region in certain cases. In these cases we observe that in most transcripts, the J region and C region overlap by exactly one base.
- Add an allele of the gene IGHJ6 to the human VDJ reference.
-
Bug fix in contig annotation: If a reference D region matches a contig perfectly, annotate the contig with that D region.
-
The command line argument
--chainis added back in 4.0 for rare cases when the automatic chain detection fails. -
A new output
airr_rearrangement.tsvis added, which contains annotated contigs of VDJ rearrangements in the AIRR TSV format. -
The VDJ reference is copied to the outputs folder starting with Cell Ranger v4.0.