Cell Ranger v7.0 and later supports analyzing Flex data with the cellranger multi pipeline (see Supported Libraries table). Flex data cannot be analyzed with the cellranger count pipeline.
This page provides specific details on setting up the multi config CSV for analyzing Universal Flex Gene Expression and Feature Barcode (Antibody Capture and CRISPR Guide Capture) libraries using the cellranger multi pipeline. Visit the List of Inputs page for a comprehensive list of all inputs required to run the Cell Ranger multi pipeline.
Cell Ranger relies on a specific multi config CSV structure for proper execution. Failure to comply with the required format (e.g., column headers, delimiters) will lead to parsing errors. Ensure that the multi config is saved in CSV format with the CSV extension.
For general information on setting up and running the multi pipeline, visit the Cell Ranger multi pipeline page. Go to the Cell Ranger Multi Config CSV page for a complete list of options for each section.
To generate a multi config CSV template, run cellranger multi-template and see the usage instructions here.
Examples of multi config CSVs for the most common library combinations are provided here. If your specific library combination is not listed and you need assistance, please contact 10x Genomics Support at support@10xgenomics.com.
Here are a few example multi config CSVs for some common Flex assay configurations, along with simplified diagrams for the corresponding experimental set up. Replace /path/to with the absolute path to your data, and customize the text according to the experiment's sample, library, and file names. Ensure that the multi config is saved in CSV format with the CSV extension.
Important: In the examples below, we set create-bam to "false" so Cell Ranger will not generate a BAM file. This setting is recommended for Flex libraries and will reduce both the total computation time for the pipestance and the size of the output directory.
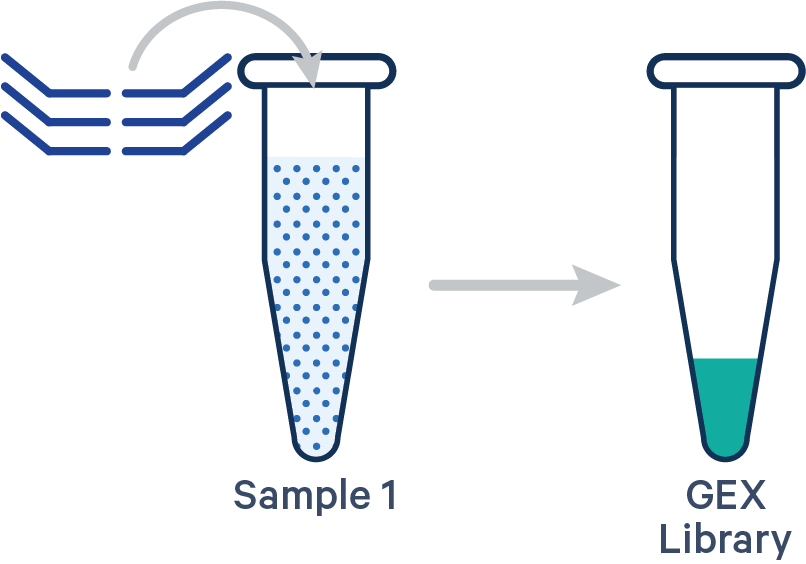
This library configuration does not use the [samples] section in the multi config CSV. The sample ID will be specified by the cellranger multi --id input. See example dataset.
Cell type annotation is enabled by setting cell-annotation-model,auto within the [gene-expression] section. For token generation and access instructions, refer to the cellranger annotate documentation.
[gene-expression]
reference,/path/to/transcriptome
probe-set, /path/to/probe-set.csv # e.g., path within your downloaded Cell Ranger tarball: cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
create-bam,false #do not generate BAM file
tenx-cloud-token-path,/path/to/10xcloud_token.json
cell-annotation-model,auto
[libraries]
fastq_id,fastqs,feature_types
flex_gex,/path/to/fastqs,Gene Expression
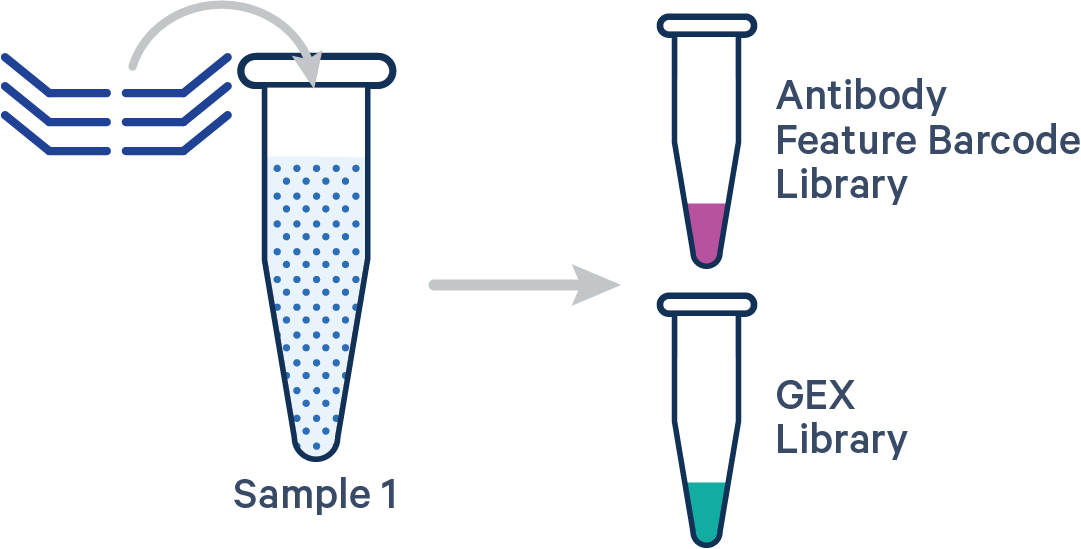
Antibody Capture is compatible in this configuration. There is one sample, one Probe Barcode, and two libraries (Gene Expression and Antibody Capture). This example applies to Antibody Capture libraries created using TotalSeq™-B, TotalSeq™-C, or Proteintech Genomics (PTG) antibodies.
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv #e.g., cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
create-bam,false #do not generate BAM file
[libraries]
fastq_id,fastqs,feature_types
flex_gex,/path/to/fastqs,Gene Expression
flex_ab,/path/to/fastqs,Antibody Capture
[feature]
reference,/path/to/feature_reference.csv
This library configuration does not use the [samples] section in the multi config CSV. The sample ID will be specified by the cellranger multi --id input. See example dataset.
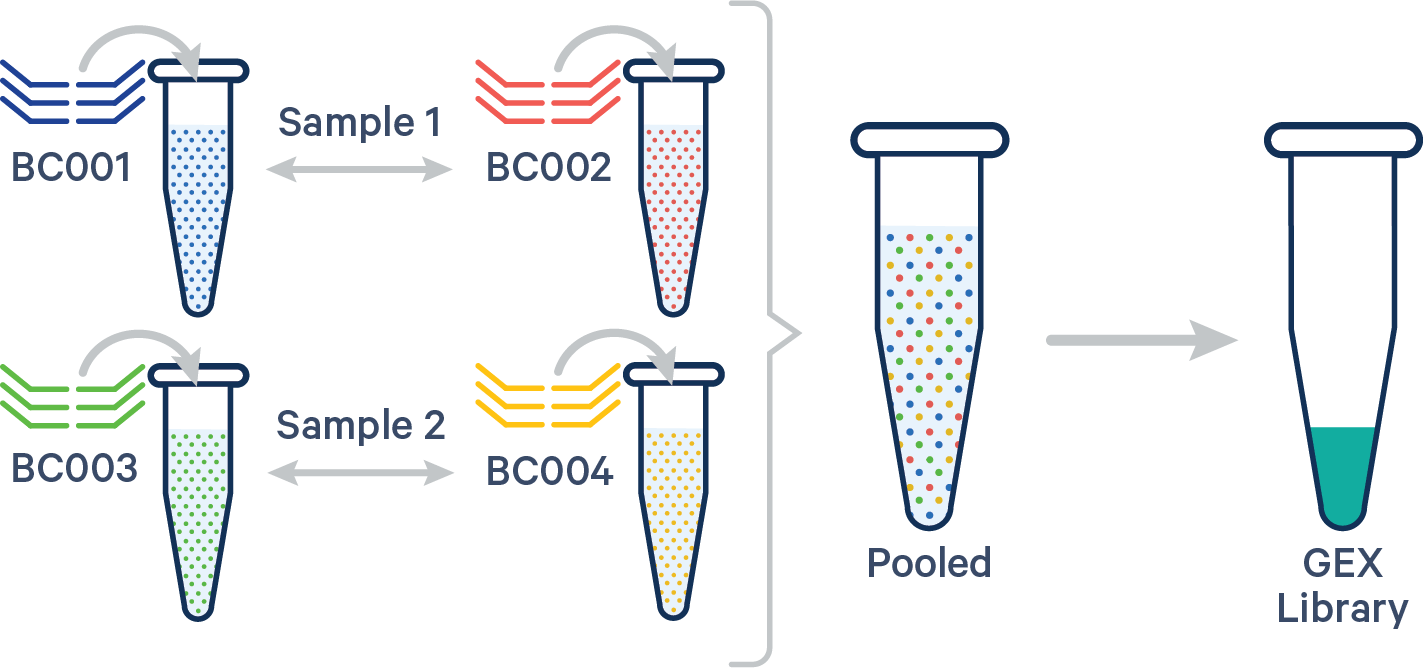
Multiple biological samples
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv #e.g., cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
create-bam,false #do not generate BAM file
[libraries]
fastq_id,fastqs,feature_types
flex_gex,/path/to/fastqs,Gene Expression
[samples]
sample_id,probe_barcode_ids,description
sample1,BC001|BC002,Control
sample2,BC003|BC004,Treated
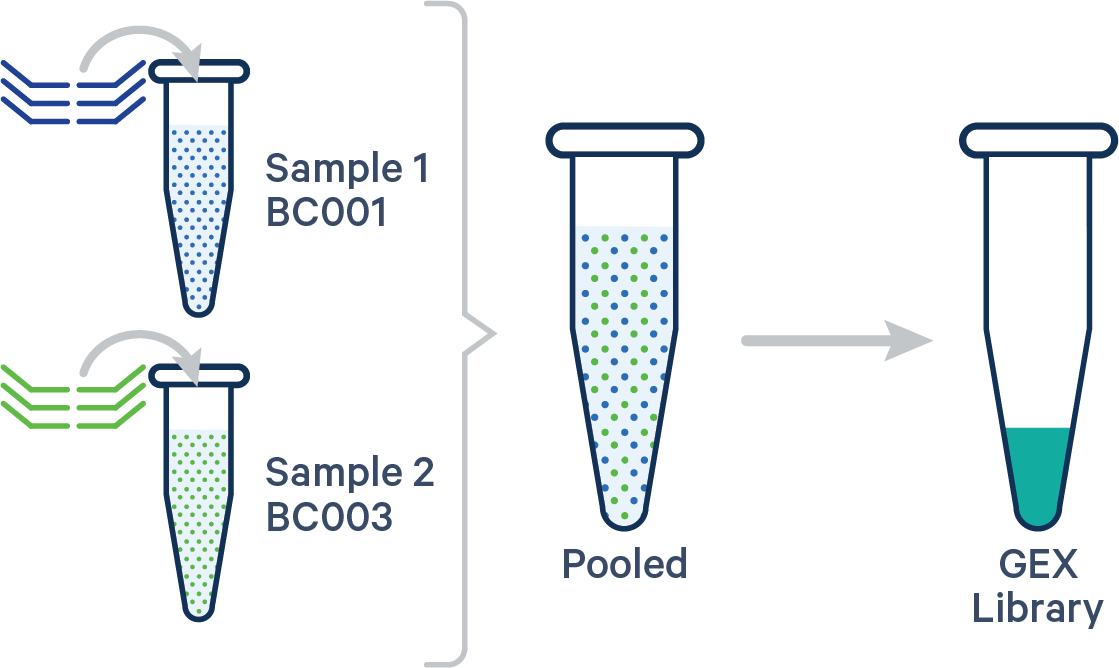
Single biological sample
In this case, the config CSV must include a [samples] section to specify the Probe Barcodes since two Probe Barcodes were used for a single sample in this experiment.
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv #e.g., cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
create-bam,false #do not generate BAM file
[libraries]
fastq_id,fastqs,feature_types
flex_gex,/path/to/fastqs,Gene Expression
[samples]
sample_id,probe_barcode_ids,description
sample1,BC001|BC002,Control
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv #e.g., cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
create-bam,false #do not generate BAM file
[libraries]
fastq_id,fastqs,feature_types
flex_gex,/path/to/fastqs,Gene Expression
[samples]
sample_id,probe_barcode_ids,description
sample1,BC001,Control
sample2,BC003,Treated
These examples apply to Antibody Capture libraries created using TotalSeq™-C or PTG antibodies.
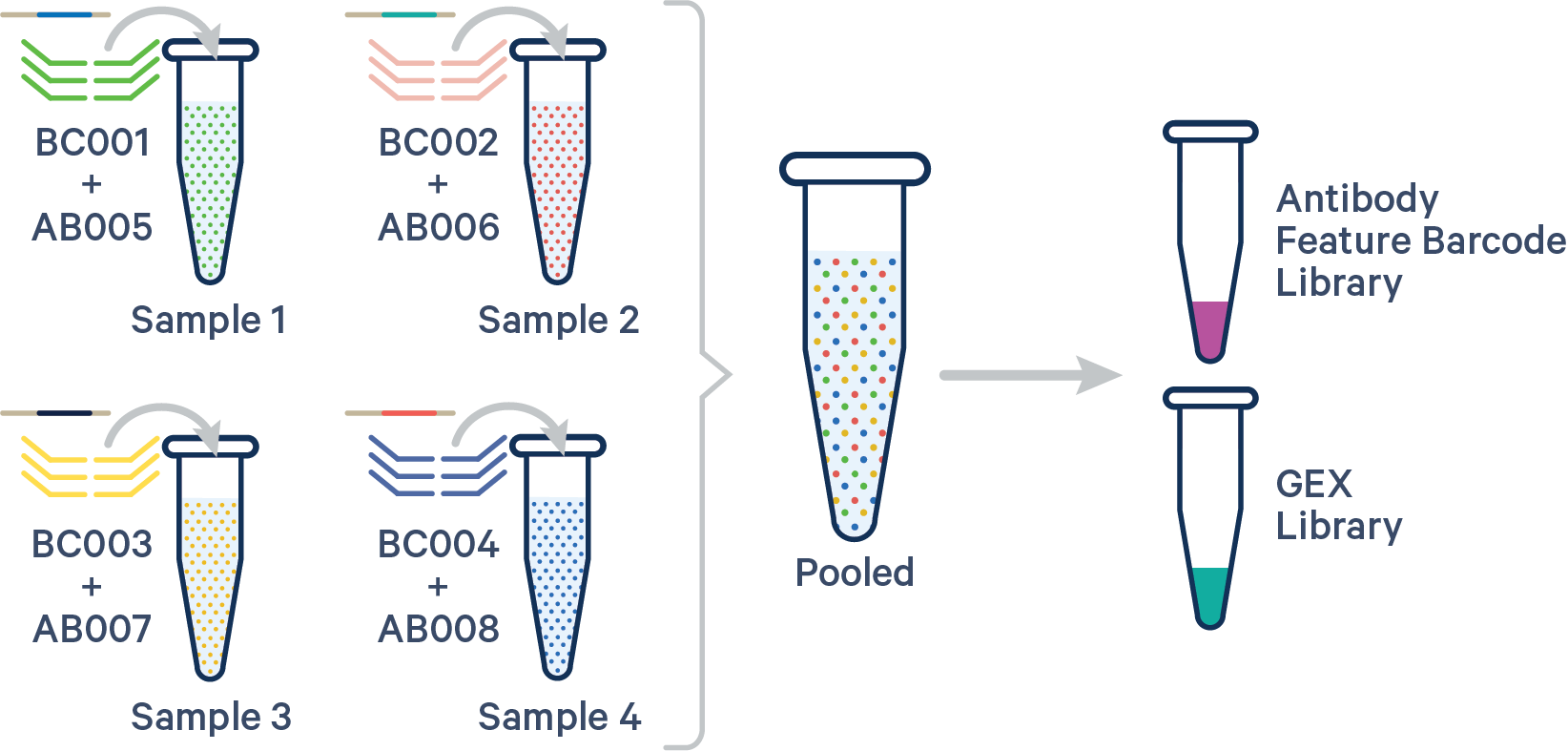
(Recommended) Specify both the Probe Barcode and Antibody Multiplexing Barcode pairs:
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv #e.g., cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
create-bam,false #do not generate BAM file
[libraries]
fastq_id,fastqs,feature_types
flex_gex,/path/to/fastqs,Gene Expression
flex_ab,/path/to/fastqs,Antibody Capture
[feature]
reference,/path/to/feature_reference.csv
[samples]
sample_id,probe_barcode_ids,description
sample1,BC001+AB005
sample2,BC002+AB006
sample3,BC003+AB007
sample4,BC004+AB008
(Advanced) Specify the Probe Barcode only, Antibody Multiplexing Barcode auto-detected:
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv #e.g., cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
create-bam,false #do not generate BAM file
[libraries]
fastq_id,fastqs,feature_types
flex_gex,/path/to/fastqs,Gene Expression
flex_ab,/path/to/fastqs,Antibody Capture
[feature]
reference,/path/to/feature_reference.csv
[samples]
sample_id,probe_barcode_ids,description
sample1,BC001
sample2,BC002
sample3,BC003
sample4,BC004
The analysis of multiplex Flex and CRISPR data is enabled in Cell Ranger v8.0 and later. For instructions on designing probes for CRISPR Guide Capture with the Flex Assay, please refer to this Knowledge Base article.
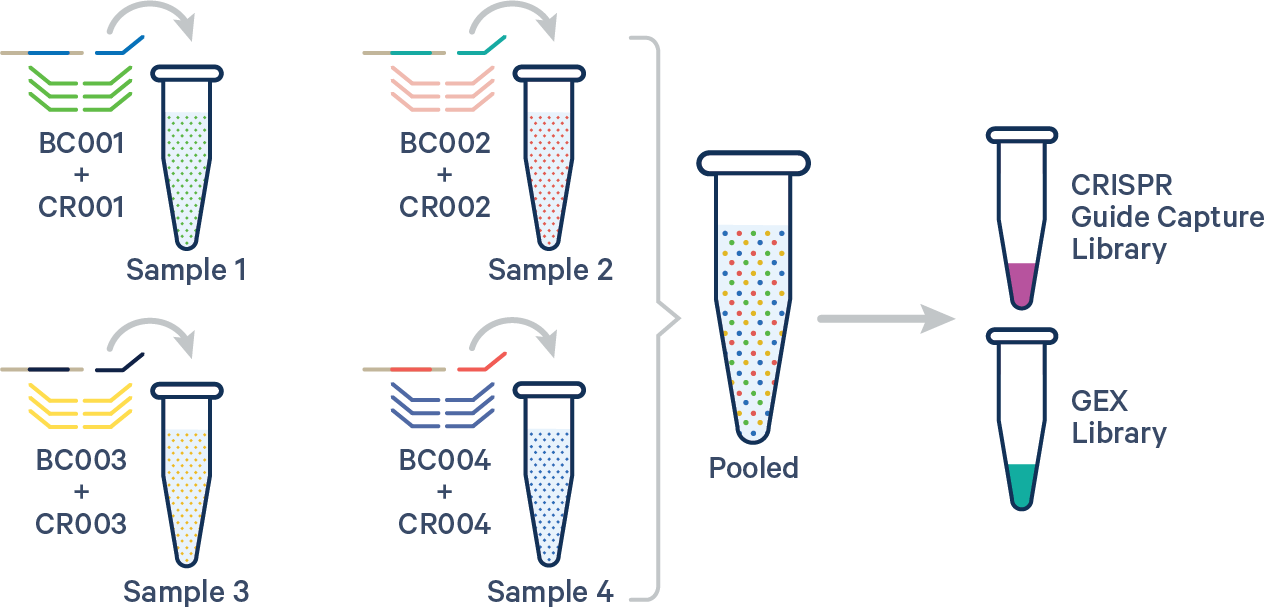
To analyze multiplex experiments, the multi config CSV must specify both the Probe Barcode and CRISPR Multiplexing Barcode, e.g., BC001+CR001 for the GEX and CRISPR libraries, respectively. Barcode auto-pairing is disabled for CRISPR Guide Capture libraries. See the example below for reference:
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv
create-bam,false
[libraries]
fastq_id,fastqs,feature_types
flex_gex,/path/to/fastqs,Gene Expression
flex_cr,/path/to/fastqs,CRISPR Guide Capture
[feature]
reference,/path/to/feature_reference.csv
[samples]
sample_id,probe_barcode_ids
sample1,BC001+CR001
sample2,BC002+CR002
sample3,BC003+CR003
sample4,BC004+CR004
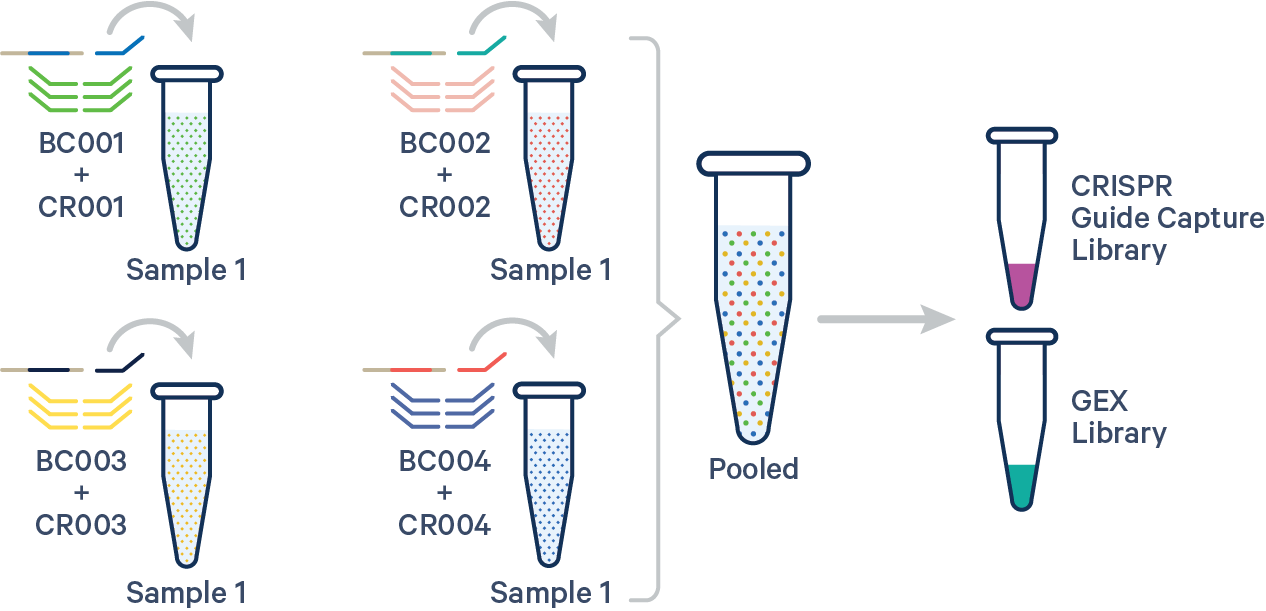
An example multi config CSV for a single biological sample with multiple Probe Barcodes and multiple CRISPR guide RNAs is shown below:
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv
create-bam,false
[libraries]
fastq_id,fastqs,feature_types
flex_gex,/path/to/fastqs,Gene Expression
flex_cr,/path/to/fastqs,CRISPR Guide Capture
[feature]
reference,/path/to/feature_reference.csv
[samples]
sample_id,probe_barcode_ids
sample1,BC001+CR001|BC002+CR002|BC003+CR003|BC004+CR004
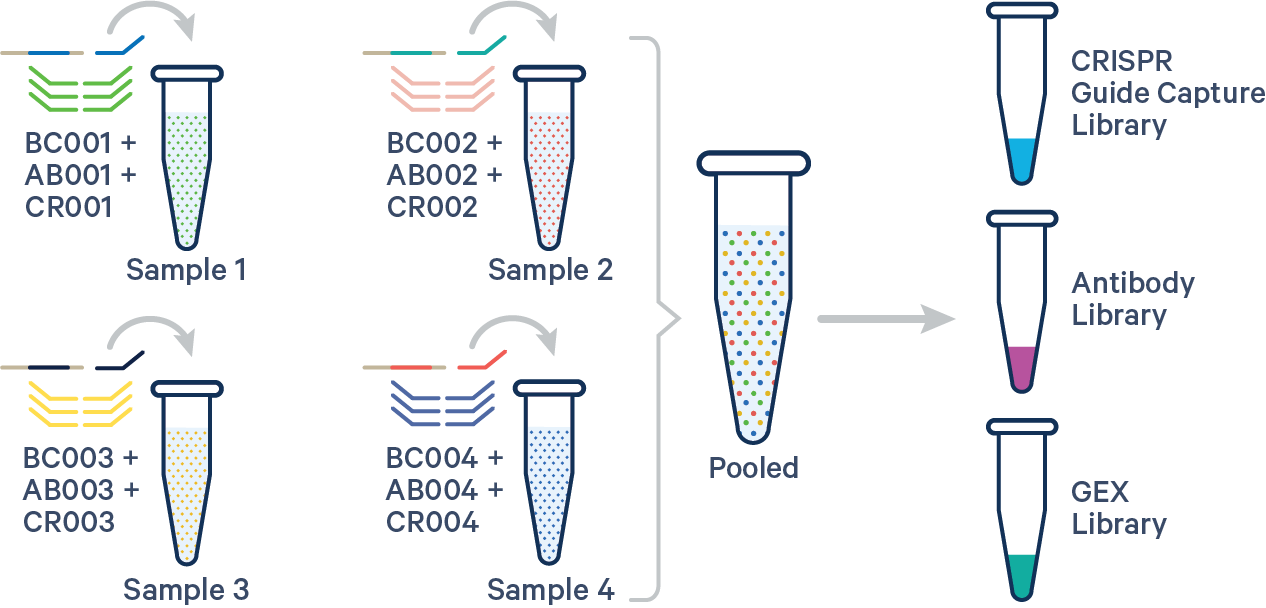
An example multi config CSV for a four biological samples with one Probe Barcodes, one Antibody barcode, and one CRISPR barcode per sample:
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv
create-bam,false
[libraries]
fastq_id,fastqs,feature_types
flex_gex,/path/to/fastqs,Gene Expression
flex_cr,/path/to/fastqs,Antibody Capture
flex_cr,/path/to/fastqs,CRISPR Guide Capture
[feature]
reference,/path/to/feature_reference.csv
[samples]
sample_id,probe_barcode_ids
sample1,BC001+AB001+CR001
sample2,BC002+AB002+CR002
sample3,BC003+AB003+CR003
sample4,BC004+AB004+CR004
The probe set reference CSV files for human and mouse are located in the probe_sets directory of the Cell Ranger package (v7.1 and later):
cellranger-9.0.0/probe_sets/
└── Chromium_Human_Transcriptome_Probe_Set_v1.1.0_GRCh38-2024-A.csv
└── Chromium_Mouse_Transcriptome_Probe_Set_v1.1.0_GRCm39-2024-A.csv
For compatibility details between Cell Ranger versions, probe set versions, and transcriptome references, see the FAQs section of the Probe Sets page.
Probe set reference CSV files and additional support files for human and mouse can also be downloaded from the Cell Ranger Downloads page. These files are described in detail on the Probe Sets Overview page.