On this page, we describe how to format custom sequence information for common Xenium v1 and Xenium Prime advanced custom targets. The files associated with these targets must be uploaded to the Xenium Panel Designer when following the advanced workflow steps to complete the request.
Guidance is provided below for the following advanced targets:
- Exogenous sequences (e.g., viral, bacterial, fluorescent reporters, or transgenes)
- Junction sequences (e.g., gene fusions and gene isoforms)
- Single nucleotide variants and indels
- CDR3 clonotypes
- Barcode detection (e.g., lineage tracing)
- CRISPR guide RNA
The Xenium Panel Designer requires FASTA files to specify advanced target sequences. Examples are provided for several advanced target types on this page below.
General information about the FASTA format for nucleotide sequences is available on the NCBI website. You can make the FASTA file using a text editor (i.e., Notepad++, VS Code, Mac terminal program such as nano).
- The sequence name line must begin with
>. - Sequences downloaded from public databases often contain multiple pieces of information in the header line that are separated by spaces. The first token of the sequence name (between the
>and the first space) will be used in downstream outputs for the gene name (i.e.,>NM_004985 KRAS-CDS-sequencewill be namedNM_004985in the final design). We suggest modifying the sequence header to include source information such as the accession number, gene name, and variant number, as applicable. Separate names with an underscore (i.e.,NM_004985_KRAS-CDS-sequence). - The DNA sequence must begin on a new line after the sequence name. The sequence can be interleaved (fixed number of characters per line) or sequential (one line).
- The sequence must be nucleotides (ATCG); protein sequences are not accepted. Nucleotides can be upper and/or lower case. XPD will convert lower case characters to upper case.
- If you are requesting multiple sequences, save them in 1 FASTA file.
- Save the file with either the
.fastaor.fafile extension. For some text editor software, you may need to change the save option to "Plain text" and edit the.txtfile extension to.fastaor.fa.
The Xenium Panel Designer requires CSV files for short variant custom targets, such as SNVs, insertions, or deletions. Examples are provided on this page below.
General information about the CSV format is available here. You can either make the CSV file using a text editor (i.e., Notepad++, VS Code, Mac terminal program such as nano) or create it in Excel.
- The file must be comma-delimited.
- There should be four columns named exactly as:
sequence,start,ref,alt(column order does not matter). - If you are requesting multiple variants, save them in 1 CSV file (new row per variant).
- Save the file with the
.csvfile extension.
Exogenous genes include protein tags, fluorescent reporters, transgenes, or expressed sequences such as CRISPR guides. All of these genes can be specified by providing a FASTA file of the sequence you would like to target where:
- This must be in the "sense" (5' to 3') orientation. This is the standard way transcripts are provided from NCBI.
- We recommend that you provide the coding sequence rather than UTRs.
- The total sequence length must be at least 80 bp.
In this guided demo, you will learn how to find exogenous gene sequence information and set up the input file for the Xenium Panel Designer. For this example, we will create a FASTA file to target two exogenous genes using information from the NCBI database. We used a search engine to find nucleotide sequence for our genes of interest. It is important the RNA sequence is exactly what you are trying to target. If the sequence is unavailable, we suggest sequencing it first.
Here is the example FASTA file for GFP and vpr:
>L29345.1_GFP
TACACACGAATAAAAGATAACAAAGATGAGTAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTT
GTTGAATTAGATGGCGATGTTAATGGGCAAAAATTCTCTGTCAGTGGAGAGGGTGAAGGTGATGCAACAT
ACGGAAAACTTACCCTTAAATTTATTTGCACTACTGGGAAGCTACCTGTTCCATGGCCAACACTTGTCAC
TACTTTCTCTTATGGTGTTCAATGCTTTTCAAGATACCCAGATCATATGAAACAGCATGACTTTTTCAAG
AGTGCCATGCCCGAAGGTTATGTACAGGAAAGAACTATATTTTACAAAGATGACGGGAACTACAAGACAC
GTGCTGAAGTCAAGTTTGAAGGTGATACCCTTGTTAATAGAATCGAGTTAAAAGGTATTGATTTTAAAGA
AGATGGAAACATTCTTGGACACAAAATGGAATACAACTATAACTCACATAATGTATACATCATGGCAGAC
AAACCAAAGAATGGAATCAAAGTTAACTTCAAAATTAGACACAACATTAAAGATGGAAGCGTTCAATTAG
CAGACCATTATCAACAAAATACTCCAATTGGCGATGGCCCTGTCCTTTTACCAGACAACCATTACCTGTC
CACACAATCTGCCCTTTCCAAAGATCCCAACGAAAAGAGAGATCACATGATCCTTCTTGAGTTTGTAACA
GCTGCTGGGATTACACATGGCATGGATGAACTATACAAATAAATGTCCAGACTTCCAATTGACACTAAAG
TGTCCGAACAATTACTAAATTCTCAGGGTTCCTGGTTAAATTCAGGCTGAGACTTTATTTATATATTTAT
AGATTCATTAAAATTTTATGAATAATTTATTGATGTTATTAATAGGGGCTATTTTCTTATTAAATAGGCT
ACTGGAGTGTAT
>NC_001802.1:5105-5396_HIV_vpr
ATGGAACAAGCCCCAGAAGACCAAGGGCCACAGAGGGAGCCACACAATGAATGGACACTAGAGCTTTTAG
AGGAGCTTAAGAATGAAGCTGTTAGACATTTTCCTAGGATTTGGCTCCATGGCTTAGGGCAACATATCTA
TGAAACTTATGGGGATACTTGGGCAGGAGTGGAAGCCATAATAAGAATTCTGCAACAACTGCTGTTTATC
CATTTTCAGAATTGGGTGTCGACATAGCAGAATAGGCGTTACTCGACAGAGGAGAGCAAGAAATGGAGCC
AGTAGATCCTAG
To target junction sequence, provide a FASTA file where:
- The sequence of each splice junction you want to target, where the splice junction is centered and there are at least 40 bases of transcribed sequence on both sides in transcription orientation.
- This must be in the "sense" (5' to 3') orientation. This is the standard way transcripts are provided from NCBI.
- The total sequence length must be at least 80 bp.
In this guided demo, you will learn how to find sequence information for isoform junctions and set up input files for the Xenium Panel Designer. For this example, we will set up an input file to specify probes for the human EGFRvIII splice variants that join exon 1 & exon 8 and exon 1 & exon 2, using information from the Ensembl database. Use the same approach to target gene fusions, where the exon sequences may come from two genes or a specific fusion sequence if available.
Here is the example FASTA file for specifying probes for the human EGFRvIII variant. We copy/pasted 60 bp of sequence for this example, as this is easier to count since it is the length of sequence in one row on the Ensembl website:
>ENST00000275493.7_EGFR_exon1_exon8
CGCTCCTGGCGCTGCTGGCTGCGCTCTGCCCGGCGAGTCGGGCTCTGGAGGAAAAGAAAGGTAATTATGTGGTGACAGATCACGGCTCGTGCGTCCGAGCCTGTGGGGCCGACAGCTATG
>ENST00000275493.7_EGFR_exon1_exon2
CGCTCCTGGCGCTGCTGGCTGCGCTCTGCCCGGCGAGTCGGGCTCTGGAGGAAAAGAAAGTTTGCCAAGGCACGAGTAACAAGCTCACGCAGTTGGGCACTTTTGAAGATCATTTTCTCA
These sequences are 120 bp in total length with the splice junction in the center.
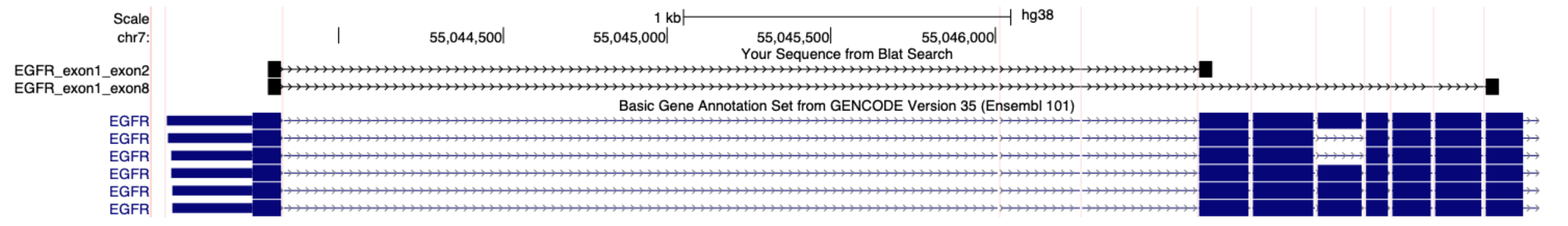
The above FASTA file is aligned to the human GRCh38 reference transcriptome. Each sequence is evenly split over the target splice junctions. Note that the sequences are in transcription orientation and only contain exonic sequence.
Short variant requests require two pieces of information:
- A FASTA file with reference sequence information.
- A CSV file with variant information. The CSV file should have four columns named exactly as:
sequence,start,ref, andalt(column order does not matter). Thestartcolumn should use 1-based position coordinates, with each alternative base in thealtcolumn (one row per alternative base).
In this guided demo, you will learn how to find sequence and variant information and set up input files for the Xenium Panel Designer. For this example, we will set up input files to target transcript variant 1 of PTEN, a tumor suppressor gene, using information from the ClinVar database. Here is an example for finding the SNV position relative to the whole transcript sequence. See the section below for finding the position relative to the CDS sequence.
Here are the first six lines of the example FASTA file for PTEN variant 1 whole transcript sequence (full file available for download here):
>NM_000314.8_PTEN_var1
GTTCTCTCCTCTCGGAAGCTGCAGCCATGATGGAAGTTTGAGAGTTGAGCCGCTGTGAGGCGAGGCCGGG
CTCAGGCGAGGGAGATGAGAGACGGCGGCGGCCGCGGCCCGGAGCCCCTCTCAGCGCCTGTGAGCAGCCG
CGGGGGCAGCGCCCTCGGGGAGCCGGCCGGCCTGCGGCGGCGGCAGCGGCGGCGTTTCTCGCCTCCTCTT
CGTCTTTTCTAACCGTGCAGCCTCTTCCTCGGCTTCTCCTGAAAGGGAAGGTGGAAGCCGTGGGCTCGGG
CGGGAGCCGGCTGAGGCGCGGCGGCGGCGGCGGCACCTCCCGCTCCTGGAGCGGGGGGGAGAAGCGGCGG
CGGCGGCGGCCGCGGCGGCTGCAGCTCCAGGGAGGGGGTCTGAGTCGCCTGTCACCATTTCCAGGGCTGG
[...]
Here is the example CSV file format. Relative to the whole transcript sequence, the start is the 1,129th base:
sequence,start,ref,alt
NM_000314.8_PTEN_var1,1129,C,T
Here is an example for finding the PTEN variant 1 SNV position relative to the CDS sequence. The CDS sequence can be found from the GenBank report page: scroll down and click on "CDS" > click on "FASTA" (bottom right corner). The CDS region for this gene is position 846 - 2057.
Here is the example FASTA file for the PTEN variant 1 CDS sequence:
>NM_000314.8:846-2057_PTEN_var1_CDS
ATGACAGCCATCATCAAAGAGATCGTTAGCAGAAACAAAAGGAGATATCAAGAGGATGGATTCGACTTAG
ACTTGACCTATATTTATCCAAACATTATTGCTATGGGATTTCCTGCAGAAAGACTTGAAGGCGTATACAG
GAACAATATTGATGATGTAGTAAGGTTTTTGGATTCAAAGCATAAAAACCATTACAAGATATACAATCTT
TGTGCTGAAAGACATTATGACACCGCCAAATTTAATTGCAGAGTTGCACAATATCCTTTTGAAGACCATA
ACCCACCACAGCTAGAACTTATCAAACCCTTTTGTGAAGATCTTGACCAATGGCTAAGTGAAGATGACAA
TCATGTTGCAGCAATTCACTGTAAAGCTGGAAAGGGACGAACTGGTGTAATGATATGTGCATATTTATTA
CATCGGGGCAAATTTTTAAAGGCACAAGAGGCCCTAGATTTCTATGGGGAAGTAAGGACCAGAGACAAAA
AGGGAGTAACTATTCCCAGTCAGAGGCGCTATGTGTATTATTATAGCTACCTGTTAAAGAATCATCTGGA
TTATAGACCAGTGGCACTGTTGTTTCACAAGATGATGTTTGAAACTATTCCAATGTTCAGTGGCGGAACT
TGCAATCCTCAGTTTGTGGTCTGCCAGCTAAAGGTGAAGATATATTCCTCCAATTCAGGACCCACACGAC
GGGAAGACAAGTTCATGTACTTTGAGTTCCCTCAGCCGTTACCTGTGTGTGGTGATATCAAAGTAGAGTT
CTTCCACAAACAGAACAAGATGCTAAAAAAGGACAAAATGTTTCACTTTTGGGTAAATACATTCTTCATA
CCAGGACCAGAGGAAACCTCAGAAAAAGTAGAAAATGGAAGTCTATGTGATCAAGAAATCGATAGCATTT
GCAGTATAGAGCGTGCAGATAATGACAAGGAATATCTAGTACTTACTTTAACAAAAAATGATCTTGACAA
AGCAAATAAAGACAAAGCCAACCGATACTTTTCTCCAAATTTTAAGGTGAAGCTGTACTTCACAAAAACA
GTAGAGGAGCCGTCAAATCCAGAGGCTAGCAGTTCAACTTCTGTAACACCAGATGTTAGTGACAATGAAC
CTGATCATTATAGATATTCTGACACCACTGACTCTGATCCAGAGAATGAACCTTTTGATGAAGATCAGCA
TACACAAATTACAAAAGTCTGA
Here is the example CSV file format. Using the information from the coding DNA reference sequence notation (c.284C>T), the SNV position relative to the start of the CDS sequence is the 284th base. As above, the reference and alternate bases are still C and T, respectively.
sequence,start,ref,alt
NM_000314.8:846-2057_PTEN_var1_CDS,284,C,T
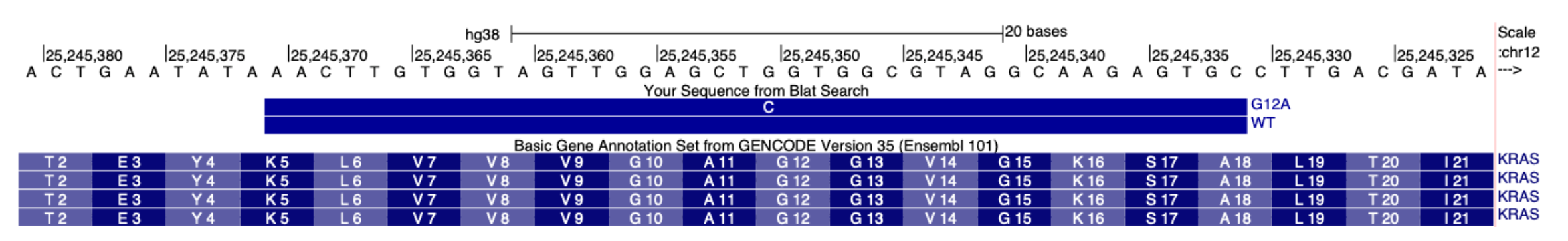
Here is an example FASTA file for specifying the KRAS G12D mutation relative to the CDS sequence:
>NM_004985_KRAS-CDS-sequence
ATGACTGAATATAAACTTGTGGTAGTTGGAGCTGGTGGCGTAGGCAAGAGTGCCTTGACGATACAGCTAA
TTCAGAATCATTTTGTGGACGAATATGATCCAACAATAGAGGATTCCTACAGGAAGCAAGTAGTAATTGA
TGGAGAAACCTGTCTCTTGGATATTCTCGACACAGCAGGTCAAGAGGAGTACAGTGCAATGAGGGACCAG
TACATGAGGACTGGGGAGGGCTTTCTTTGTGTATTTGCCATAAATAATACTAAATCATTTGAAGATATTC
ACCATTATAGAGAACAAATTAAAAGAGTTAAGGACTCTGAAGATGTACCTATGGTCCTAGTAGGAAATAA
ATGTGATTTGCCTTCTAGAACAGTAGACACAAAACAGGCTCAGGACTTAGCAAGAAGTTATGGAATTCCT
TTTATTGAAACATCAGCAAAGACAAGACAGGGTGTTGATGATGCCTTCTATACATTAGTTCGAGAAATTC
GAAAACATAAAGAAAAGATGAGCAAAGATGGTAAAAAGAAGAAAAAGAAGTCAAAGACAAAGTGTGTAAT
TATGTAA
Here is the example CSV file format. Using the information from the coding DNA reference sequence notation (c.35G>A), the SNV position relative to the start of the CDS sequence is the 35th base. The alternate base is an A instead of a G:
sequence,start,ref,alt
NM_004985_KRAS-CDS-sequence,35,G,A
An example of the designed probes aligned to the human GRCh38 reference transcriptome is shown below:
Provide a FASTA file with exonic sequence. Here is an example:
>indel
ATGCATGCATGCATGCATGCATGCATGCATGCATGCATGCGGTCTCGATGTTGTCAATATTCCCCCAAGAACCCTTCTGGACAATGCATGCATGCATGCATGCATGCATGCATGCATGCATGC
>large_deletion
agagagccttgaggaaaaccaGCGGAACCTCCTTCAGATGACTGAAAAGTTcttccatgccatcatcagttcctaagggccttaccccatgcc
Additionally, provide a CSV file to define insertions and deletions. The alternative sequence should be left-padded (use the nucleotide before the event as the start, see examples below) and the positions should be 1-based. For example, the CSV file should look like this for the three scenarios listed below - short insertion, short deletion, and large deletion:
sequence,start,ref,alt
indel,43,T,TAA
indel,42,GT,
large_deletion,22,GCGGAACCTCCTTCAGATGA,
Here are three scenarios illustrating how 10x will design the probes for Xenium v1 using the example FASTA and CSV above:
-
Short insertion: This adds an AA at position 43. We left-pad the sequence with TAA, which results in this design:
seq: GCATGCATGCATGCATGCGGT CTCGATGTTGTCAATATTCC WT probe: GCATGCATGCATGCATGCGGT CTCGATGTTGTCAATATTC ALT probe: CATGCATGCATGCATGCGGTAACTCGATGTTGTCAATATT -
Short deletion: This deletes a GT. The
altcolumn is empty.seq: TGCATGCATGCATGCATGCGGTCTCGATGTTGTCAATATTCC WT probe: CATGCATGCATGCATGCGGTCTCGATGTTGTCAATATTCC ALT probe: TGCATGCATGCATGCATGCG CTCGATGTTGTCAATATTCC -
Large deletion: This removes 30 bp (upper case represents the deletion region). The
altcolumn is empty.seq: agagagccttgaggaaaaccaGCGGAACCTCCTTCAGATGACTGAAAAGTTcttccatgccatcatcagttc WT probe: gagagccttgaggaaaaccaGCGGAACCTCCTTCAGATGA ALT probe: gagagccttgaggaaaacca cttccatgccatcatcagtt
To target known CDR3 sequences, provide a FASTA file containing the CDR3 where:
- The total sequence length must be at least 80 bp.
- If the sequence is not 80 bp, additional flanking sequence from framework regions (FWR3/FWR4) can be included to ensure an optimal probe is picked. The CDR3 sequence must be in the center of the overall sequence.
- The sequence must be in "sense" (5' to 3') orientation.
For example, this representative PBMC dataset contains assembled CDR3 sequences that range from 42 bp to 48 bp. Here is an example assembled clonotype:
| Type | Sequence |
|---|---|
| Barcode | TATCAGGCACGCGAAA-1 |
| FWR3 | ACTGACCAAGGAGAAGTCCCCAATGGCTACAATGTCTCCAGATCAACCACAGAGGATTTCCCGCTCAGGCTGCTGTCGGCTGCTCCCTCCCAGACATCTGTGTACTTC |
| CDR3 | TGTGCCAGCAGCCGGGACAGGGTAAATCAGCCCCAGCATTTT |
| FWR4 | GGTGATGGGACTCGACTCTCCATCCTAG |
Converting this to a FASTA file, the inputs would look like this using 20 bp of each framework region:
>Clonotype1
CCCAGACATCTGTGTACTTCTGTGCCAGCAGCCGGGACAGGGTAAATCAGCCCCAGCATTTTGGTGATGGGACTCGACTCTC
To target barcodes, provide a FASTA file where:
- The total length of the barcode and flanking sequence should be at least 80 bp. The total barcode sequence length must be 40 bp (Xenium v1) or 60 bp (Xenium Prime 5K).
- If your barcode is shorter than 80 bp, additional non-unique sequence can be included. The designed probes will be checked for potential cross-binding.
- The sequence must be in "sense" (5' to 3') orientation.
Here is an example of three 40 bp (upper case) barcode sequences with 20 bp (lower case) of constant sequence flanking the barcode:
>Barcode_sequence_1
atgcgtacgtagctagctagATCTTCGGCGGAAACTGAGCCAGCATTACAACGTTTTCAGatgcgtacgtagctagctag
>Barcode_sequence_2
atgcgtacgtagctagctagCTGTTCCTGTTGAGGTCTAAAATATCACTTGCAGGTAGTGatgcgtacgtagctagctag
>Barcode_sequence_3
atgcgtacgtagctagctagTGCTGCTCACCTACAGTTCACCCCCAAATACCCGACCGAGatgcgtacgtagctagctag
To target CRISPR guide RNA, provide a FASTA file where:
- The total sequence length is at least 80 bp.
- The protospacer (or other uniquely targetable sequence) must be included.
- The sequence must be in "sense" (5' to 3') orientation.
Here is an example:
>gRNA1_TargetGeneX
GAGTCCGAGCAGAAGAAGAAGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTT
>gRNA2_TargetGeneY
GTGCTGACCCGAGGTCTGCTGGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTT