Mechanisms of action and resistance to epigenetic cancer therapy valemetostat, revealed with single cell ATAC-seq

Successful clinical trials take many forms. Some identify transformative therapeutics, while others fail to meet primary endpoints but yield key biological insights to guide future drug discovery efforts. In our Clinical trials to watch blog series, we feature innovative applications of single cell multiomics and spatial gene expression to clinical samples across trial phases.
Follow the series to learn how our tools enable researchers to analyze patient samples collected throughout a trial and uncover rare cell populations predictive of therapeutic responses, biomarkers to improve patient stratification, or novel mechanisms of action.
Researchers partially revealed the cellular mechanisms driving efficacy and resistance to a novel cancer therapy—that is approved for treatment of adult T-cell leukemia/lymphoma (ATL) in Japan—that targets epigenetic regulatory proteins through analysis of clinical trial samples with the Chromium Single Cell Gene Expression, Single Cell ATAC (Assay for Transposase Accessible Chromatin), and Multiome ATAC + Gene Expression assays (1).
Epigenetic regulators are an emerging therapeutic target for a variety of cancers. Many cancer cells exhibit dysregulated gene expression due, in part, to silencing of tumor suppressor genes by hypermethylation of gene promoter regions. For example, excessive H3 lysine trimethylation (H3K27me3) is considered an epigenetic hallmark of cancer.
Researchers have previously demonstrated that people with blood cancer respond positively to therapies that reduce H3K27me3.
“Before H3K27me3-inhibitor therapies were developed, no effective treatments for blood cancers with accumulated genetic abnormalities existed, and new treatments needed to be developed,” said Makoto Yamagishi, first author of the recent Nature paper and an associate professor at the University of Tokyo (2).
But cancers often become resistant to these inhibitors. Since the mechanisms of efficacy and resistance in tumor cells were poorly understood, researchers and clinicians struggled to improve the long-term efficacy of this emerging class of cancer therapeutics.
Scientists from the University of Tokyo, University of Ryukyus, and Daiichi Sankyo Co. worked together to delineate the mechanism of their first-of-its-class dual inhibitor, valemetostat, by examining chromatin accessibility and gene expression at single cell resolution. Their study provides critical insight into next-generation epigenetic therapies and an improved outlook for the treatment of the aggressive, refractory blood cancer ATL.
Phases I and II: Dual inhibition of histone modifying proteins reduces tumor growth in people with T-cell lymphoma
Cancer type: T-cell leukemia/lymphoma
Therapy: Valemetostat (DS-3201)
Trial aim: To evaluate the sustained safety and efficacy of dual inhibition of histone modifying proteins EZH1 and EZH2 in participants with relapsed or refractory non-Hodgkin’s lymphoma, including ATL
Leading institutions: University of Tokyo, University of Ryukyus
Pharmaceutical collaborators: Daiichi Sankyo Co., Ltd.
ClinicalTrials.gov registration: Phase I (NCT02732275) and Phase II (NCT04102150)
What epigenetic regulatory proteins does valemetostat target?
Valemetostat targets both methyltransferases enhancer of zeste homolog 1 (EZH1) and EZH2. Either enzyme can act as the catalytic subunit of the polycomb repressive complex 2 (PRC2)— which mediates methylation of H3K27—but they have different expression patterns, levels of enzymatic activity, and DNA/nucleosome binding activity (3–5). PCR2–EZH2 is the canonical form of the complex and better characterized than PCR2–EZH1 in both development and cancer biology. Although researchers are still delineating the distinctive functions of these complexes, some observed that PCR2–EZH1 can compensate for PCR2–EZH2. The researchers cited this as a primary reason dual inhibition is needed.
Are EZH1 and EZH2 altered in tumor cells?
Increased H3K27me3 is a hallmark of cancer, making the canonical PCR2–EZH2 complex a primary drug target of interest for a variety of cancers. EZH2-activating mutations are commonly observed in some cancers, leading to the development of EZH2 inhibitors such as tazemetostat, which is FDA-approved for treating relapsed or refractory follicular lymphoma patients with an Ezh2 mutation (6). Mutations in the Ezh1 gene in tumor cells are less common, though they have been reported in some cancers, including thyroid cancers (5). Because of this, researchers had not developed EZH1 inhibitors or EZH1/EZH2 dual inhibitors before valemetostat.
Clinical results
People with aggressive forms of ATL are often resistant to therapies commonly used for ATL—including multiagent chemotherapy and allogeneic hematopoietic stem cell transplant. Those who are responsive often relapse or develop therapeutic resistance following treatment. Reagents approved for refractory or relapsed ATL in Japan—where the virus that causes ATL is endemic—still have low response rates in people with aggressive disease.
The researchers recruited 25 patients with relapsed, recurrent, or refractory disease previously given other treatments. They achieved the primary outcome of the trial, reporting an objective response rate among patients of 48%, a complete response rate of 20%, and a partial response rate of 28% (7). The research group also reported in both their phase I and II trial results that the new therapeutic had an acceptable safety profile (7,8).
Ten participants from both phase I and phase II trials participated in additional molecular testing throughout the study, safely sustaining treatment for over two years (1). The researchers collected blood samples from the participants immediately before and during treatment. Genome sequencing of samples revealed that ATL cells with high mutational burden and profiles associated with high malignancy were significantly reduced following valemetostat treatment.
They also analyzed patient samples with chromatin immunoprecipitation (ChIP) to measure how treatment affected H3K27me3 levels in the tumor cells. Samples collected after treatment had significantly lower H3K27me3 levels. Specifically, methylation was reduced in the transcription start sites of key tumor suppressor genes and H3K27ac—an activating epigenetic modification—increased, which restored expression of these genes to levels similar to healthy cells based on bulk RNA-seq analysis.
Applications of single cell sequencing
Surveying chromatin accessibility to define the mechanism of disease and drug action
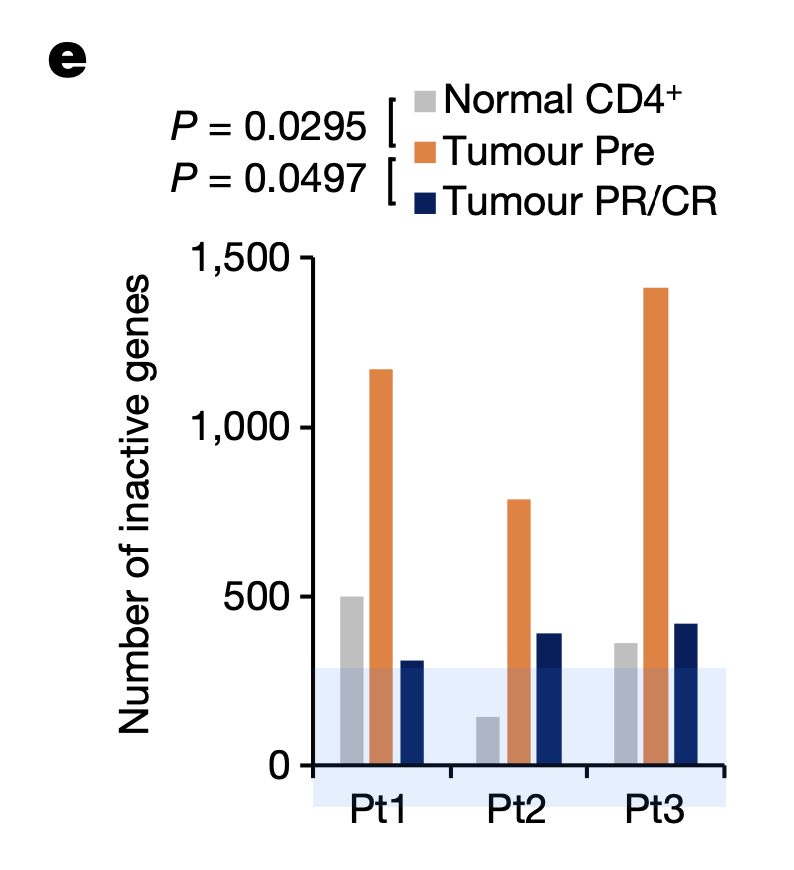
To understand the basis of efficacy for valemetostat, the research team used Chromium Single Cell ATAC (Assay for Transposase Accessible Chromatin) to explore how the drug affected chromatin structure in tumor cells. Taking PBMCs from 10 patients before treatment, at the time of response, and during disease progression, the researchers observed a condensed chromatin pattern in pre-treatment tumor cells. H3K27me3 accumulated in these condensed chromatin regions. After valemetostat treatment, however, H3K27me3 levels decreased, and the team observed a relaxation of the chromatin landscape to a level comparable to normal CD4+ T cells. Chromatin relaxation notably reduced the number of inactivated genes as well (Figure 1), including genes involved in anti-tumor T-cell function and TSGs (tumor suppressor genes).
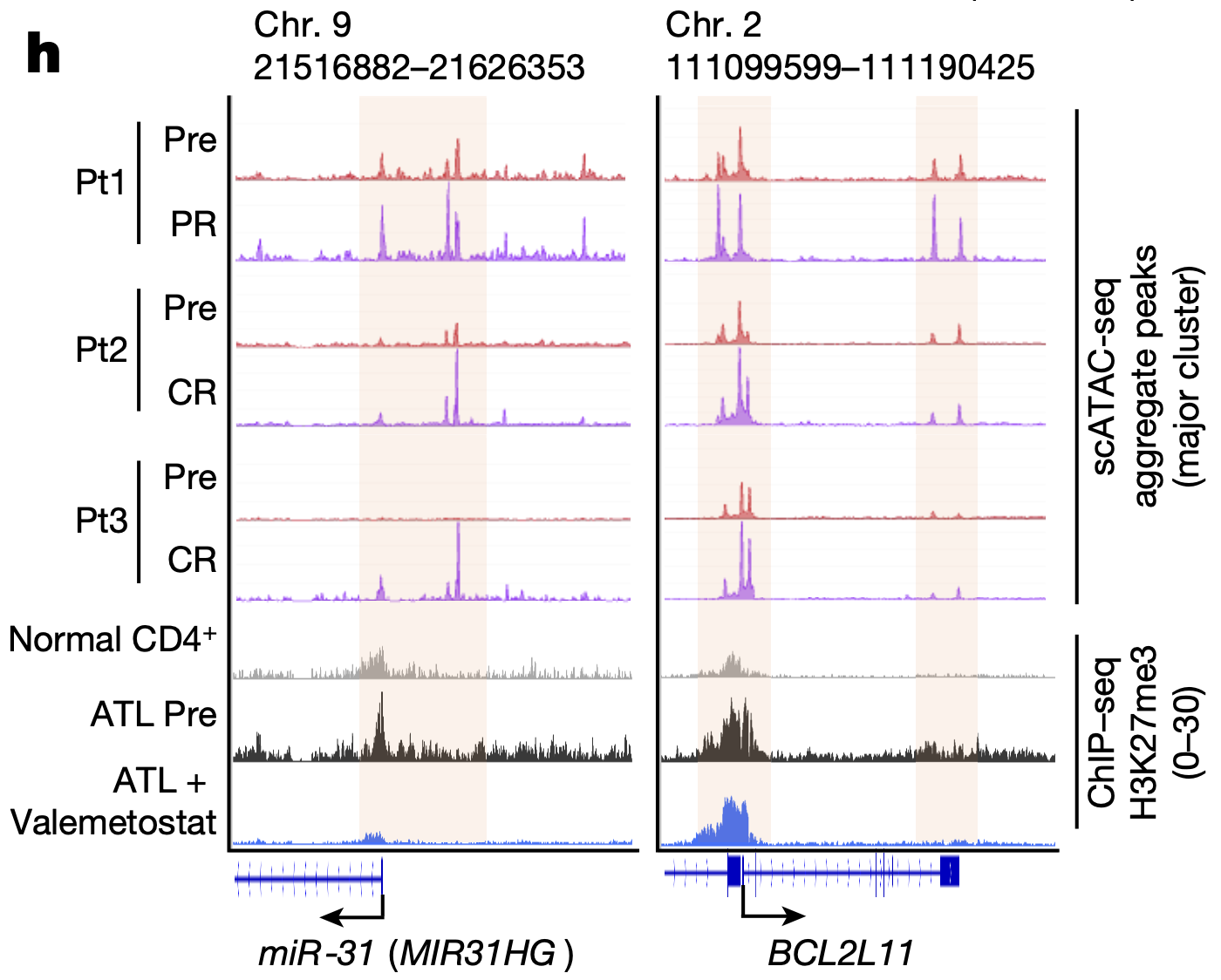
The team then used Chromium Single Cell Gene Expression to further investigate the transcriptional effects of valemetostat-induced changes in chromatin accessibility. They performed scRNA-seq on PBMCs from the same 10 patients, then integrated the data with scATAC-seq data. They found that, as valemetostat countered H3K27me3 activity, relaxing the chromatin structure, there was increased expression at the gene loci where H3K27me3 had previously accumulated, including H3K27me3 target genes miR-31 and BCL2L11 (Figure 2), which play variable functions in human cancer. Additionally, their single cell transcriptional insights revealed that 246 genes were upregulated in tumor cells before treatment, including genes involved in cell growth and apoptosis regulation; however, 89.4% of these abnormal genes were subsequently repressed through the epigenomic regulations caused by valemetostat.
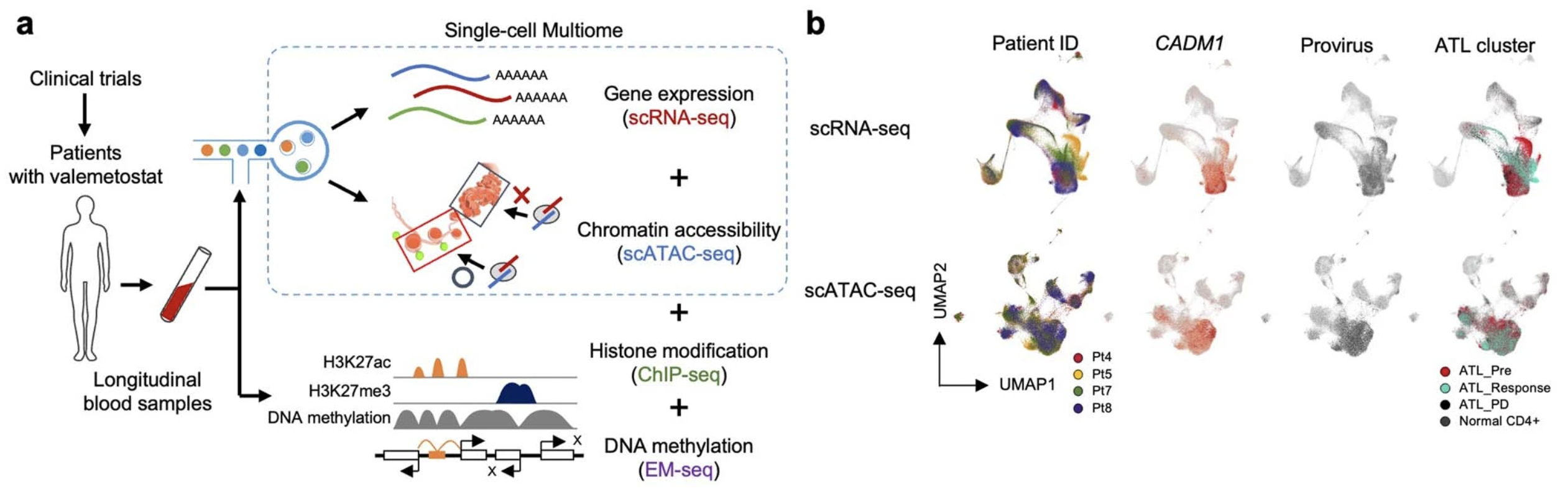
Sampling PBMCs from the validation phase II cohort, which included four patients, the team confirmed insights from their phase I trial results using Chromium Single Cell Multiome ATAC + Gene Expression (Figure 3). They analyzed 109,830 cells, obtaining ATAC and gene expression data from the same individual cells, and observed that changes in chromatin structure were responsible for gene expression programs driving tumorigenesis; and, in contrast, valemetostat treatment did counter H3K27me3-meditated chromatin aggregation and gene silencing.
Together these findings define a mechanism of tumorigenesis that hinges on oncogenic expression programs and H3K27me3 activity to condense chromatin. Valemetostat has opposing effects, relaxing chromatin structures and subsequently activating crucial gene expression programs involved in the anti-tumor immune response, while deactivating oncogenes.
Understanding the clonal basis of ATL resistance and relapse
The research team continued to explore the long-term efficacy of valemetostat with single cell technologies, particularly after the recurrence of ATL in patients who had originally shown durable responses to the treatment. What was the basis of disease progression after an initially complete or partial response?
Insights from other technologies first confirmed the presence of somatic mutations driving resistance and tumor repopulation. Looking at tumor cells in a patient (Patient 1) from the original phase I cohort at the point of disease progression, the team identified a somatic mutation at the Y111 amino acid residue of EZH2—one of the methyltransferase enzymes that mediates methylation of H3K27 and a valemetostat target. Among five of the ten participating patients, additional somatic mutations were found in the core components of the Polycomb repressive complex 2 (PRC2), which seemed to affect the valemetostat-binding pocket, likely reducing valemetostat binding affinity to the PRC2–EZH2 complex. These mutations emerged in the original T-cell clones that were present in patients before treatment. These mutations also affected H3K27me3 levels; using protein electrophoresis, they found that 293 T cells with the mutations, though in the presence of valemetostat, had retained H3K27me3 levels similar to that of untreated tumor cells.
The team then investigated the effects of the mutations on chromatin accessibility. Using single cell ATAC-seq data from tumor cells collected after disease recurrence in two patients, they observed a return to the condensed chromatin state and resuppression of genes that had been activated by valemetostat treatment. Moreover, single cell RNA-sequencing data confirmed the expression of the mutant EZH2 in the progressive disease clone. This confirmed the mechanism of resistance caused by the somatic mutations: a complete reversal of valemetostat activity through chromatin recondensation.
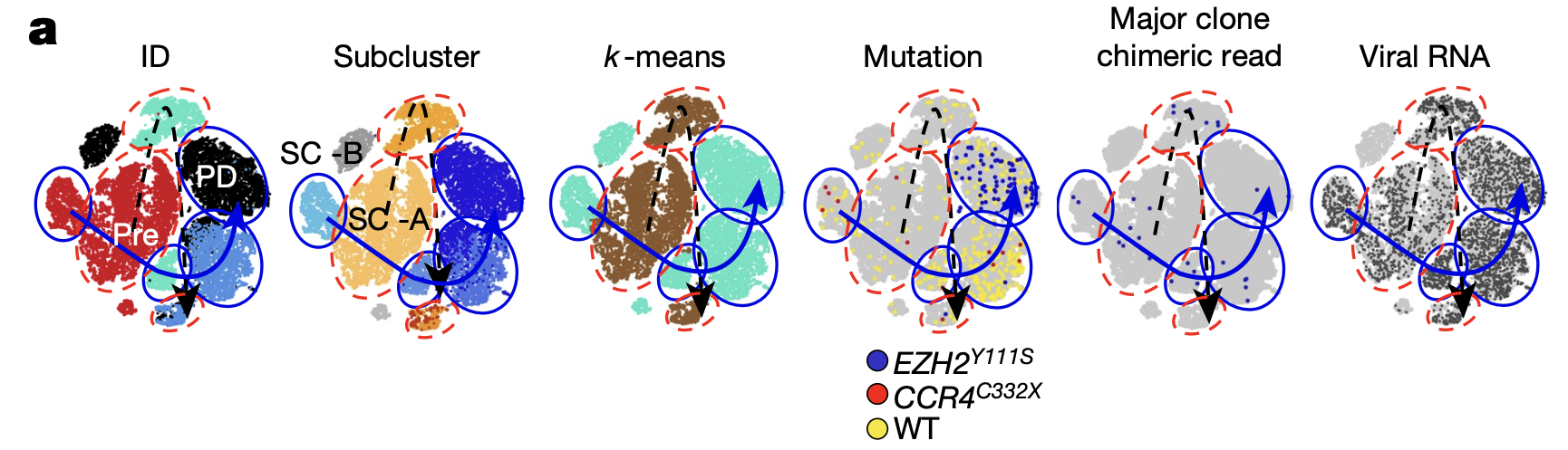
Single cell RNA-sequencing data from Patient 1 also revealed the likely T-cell clone behind progressive disease. Reclustering the single cell data taken at all time points from this patient, including that from T cells remaining after treatment, the team found two distinct pre-treatment T-cell subclusters, subcluster A and B (SC-A, SC-B) (Figure 4). Analysis of the proportional changes in these subclusters over the clinical time course showed that SC-B was infrequent before treatment and at treatment response. However, the T-cell clone that eventually acquired the somatic mutations driving resistance and subsequently expanded after treatment shared the same mutation patterns as SC-B, suggesting this subcluster represented the clone behind disease resistance and relapse.
In this study, the wealth of information provided by single cell analysis of chromatin accessibility and transcriptional changes over the course of treatment, in partnership with other powerful technologies, established a more complete understanding of ATL, valemetostat mechanism of action, and the basis for recurrence. For this challenging blood cancer, and other cancer types, these findings represent an important advancement on the path toward more effective, durable epigenetic cancer therapies.
Read the full publication detailing single cell analysis of samples from participants in the phase I and phase II clinical trials testing safety and efficacy of valemetostat.
Learn more about the technology that supported these clinical findings:
- Chromium Single Cell Gene Expression
- Chromium Single Cell ATAC
- Chromium Single Cell Multiome ATAC + Gene Expression
References:
- Yamagishi M, et al. Mechanisms of action and resistance in histone methylation-targeted therapy. Nature 627: 221–228 (2024). doi: 10.1038/s41586-024-07103-x
- Krisher J. Researchers reveal mechanism of drug reactivating tumor suppressors. EurekAlert! (2024).
- Margueron R, et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 32: 503–18 (2008). doi: 10.1016/j.molcel.2008.11.004
- Shen X, et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell 32: 491–502 (2008). doi: 10.1016/j.molcel.2008.10.016
- Lee SH, et al. The role of EZH1 and EZH2 in development and cancer. BMB Rep 55: 595–601 (2022). doi: 10.5483/BMBRep.2022.55.12.174
- Morschhauser F, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol 21: 1433–1442 (2020). doi: 10.1016/S1470-2045(20)30441-1
- Izutsu K, et al. An open-label, single-arm phase 2 trial of valemetostat for relapsed or refractory adult T-cell leukemia/lymphoma. Blood 141: 1159–1168 (2023). doi: 10.1182/blood.2022016862
- Morishima S, et al. First-in-human study of the EZH1/2 dual inhibitor valemetostat in relapsed or refractory non-Hodgkin lymphoma (NHL) - Updated results focusing on adult T-cell leukemia-lymphoma (ATL). Blood 134(Supplement_1): 4025 (2019). doi: 10.1182/blood-2019-125507
About the authors:


Blog contributor: This article has been reviewed for scientific accuracy by Angela Churchill, PhD.
