How high-definition spatial transcriptomics, single cell FFPE, and Xenium are tackling the cancer problem showcased at AACR 2024

In her opening plenary session at AACR 2024, Aviv Regev, PhD, Head of Genentech Research and Early Development, pointed out the huge problem—indeed, the potentially overwhelming problem—of cancer. There are > 100 different cancer types, 10,000 known driver mutations, complex circuits driving cancer biology within and between cells in the tumor microenvironment, and an almost unfathomable number of possible small molecules and antibody structures to consider for drug development.
Yet, there is hope that gains can be made against this deadly disease that seems to always present big challenges and small odds to the research community, doctors, and patients. As Dr. Regev’s talk exemplified, growing cancer cell atlases and increasingly intelligent computational models are helping researchers understand patterns in the disease biology that can lead to faster, more effective cancer drug development. Perhaps, a 22,000+ person attendance, the largest in AACR history, is a testament in itself to the increasing pace of cancer research and the promise of therapeutic advancements. The world’s brightest and most driven scientists are facing the problem of cancer head-on, and leveraging comprehensive toolkits, including high-resolution single cell and spatial genomics, with powerful algorithms to work harder and smarter against cancer.
We were so proud to see 10x Genomics tools play a part in the work of these incredible scientists, including their efforts to understand the earliest stages of cancer development, identify putative biomarkers associated with clinical outcomes, and demystify the spatial organization of the tumor microenvironment. In the following sections, we highlight some of our favorite talks from AACR 2024 and how 10x Genomics technologies were used. We acknowledge that these studies are just a representation—though they are especially important to us—of a much larger story of continuous technology innovation, dedicated research, and breakthrough discoveries across the field, which AACR 2024 so powerfully captured.
Jump ahead to different featured presentations with the links below:
- Single cell RNA-seq identifies Midkine as a putative biomarker for breast cancer severity
-
Leaders discuss the intersection of spatial biology and machine learning for tumor microenvironment research
- FFPE access enables comprehensive single cell and spatial study of premalignant tissue in lung cancer
Single cell RNA-seq helps to identify a biomarker for breast cancer risk and severity in younger women
Intuition and experience suggests that older individuals are at higher risk of cancer development and negative clinical outcomes, and, in breast cancer, the data confirms it. Kornelia Polyak, MD, PhD, Professor of Medicine at Dana-Farber Cancer Institute, Harvard Medical School, and a co-leader of the Dana-Farber Harvard Cancer Center Cancer Cell Biology Program—and a world leader in breast cancer research—demonstrated that individuals over 75 are at the highest risk of disease-specific death.
However, she also introduced the AACR audience to the surprising data that the youngest individuals (< 40 years old) with breast cancer have a similar trend of risk for disease-specific death. Age thus acts as an uncertain variable in the onset and progression of breast cancer.
Dr. Polyak set out to define the age-related mechanisms underlying tumorigenesis using a mammary tumor model in rats. First interrogating healthy aging mammary tissue by histology and single cell RNA-sequencing, she and her team characterized distinct cell-type transitions between samples from younger and older rats. The B-cell and luminal-epithelial compartments decreased with age, which was also observed in existing human breast single cell datasets (1). Zooming in on changes in the mammary epithelium (the cell class that breast cancer is derived from) with age, they noted a decline in mature luminal and luminal progenitor cells and an increase in a unique “aging luminal progenitor” cell type. Gene enrichment analysis based on single cell transcriptional data showed that this aging luminal progenitor population had a decline in tumor suppressive pathways, suggesting a shift to a more cancer-permissive cell state.
Analysis of basal–luminal (two prominent epithelium cell types in mammary glands) cell ligand receptor interactions showed that the basal ligand Midkine (Mdk) was an increasingly prominent mediator of basal–luminal communication with age. Immunofluorescence staining in tumor samples further confirmed increased diffusion of Mdk expression in invasive ductal carcinoma, compared to earlier stages of breast cancer, and that Mdk expression increased with age.
Bringing this finding into a clinical context, Dr. Polyak noted in an existing RNA-seq dataset from a cohort of ER+ breast cancer patients that older women have the same trend of disease-free survival regardless of Mdk expression levels; however, higher Mdk expression in younger women was associated with shorter disease-free survival. This would indicate Mdk could represent a prognostic biomarker associated with poor outcomes in younger breast cancer patients.
To further validate this discovery, Dr. Polyak’s team treated young rats with Midkine and a control vehicle prior to establishing a breast cancer tumor model. Single cell analysis of mammary gland tissue demonstrated that basal and luminal cells had higher expression of Ki67, a gene involved in proliferation, as a result of Midkine treatment. This suggests Midkine may help to drive epithelial proliferation. Interestingly, Midkine-treated mammary tissue had strikingly similar features to the single cell transcriptomic profiles seen in mammary samples from older rats, including common upregulated/downregulated genes and the presence of the unique aging luminal progenitor cell population, indicating Midkine treatment mimics aging-related transcriptomic changes.
This detailed single cell study not only unravels some of the uncertainty around how aging affects mammary tissues, making epithelial cell types more permissive to cancer, but also points to an underlying mechanism that may explain why the youngest breast cancer patients share similar disease progression trends with the oldest.
Back to topUncovering deeper layers of biology in the tumor microenvironment through spatial tools and machine learning
A recurring theme at AACR 2024 was the role of computation—including modeling software and algorithms that train machine learning models—to derive patterns from high-resolution, high-content data (including single cell and spatial data) that, in turn, supply insights and predictions of clinical relevance. These combined capabilities seemingly represent the leading edge of cancer research as powerful wet-lab technology innovations meet equally powerful computational advancements.
A group of leaders in spatial biology took the opportunity at AACR to highlight the potential of these approaches when investigating the composition and organization of the tumor microenvironment. Their presentations provide insight into what the next era of discovery into these highly complex tissue systems could look like—moving beyond ever-crucial cellular annotations towards deeper characterization of molecular changes resulting from tumor–immune cell interactions, pathways associated with resistance, and potential predictive indicators of tumor responses to therapy.
While a number of fascinating talks touched on the value of spatial transcriptomics technology and emerging methods for computational analysis, we highlight presentations from Dr. Elana Fertig of Johns Hopkins University School of Medicine, Dr. Peter Sorger of Harvard Medical School, and Dr. Joakim Lundeberg of SciLifeLab and KTH Royal Institute of Technology in the sections below:
From spatial transcriptomics of patient tumor samples to predictive medicine
For Dr. Elana Fertig, Professor of Oncology and Division Director of Oncology Quantitative Sciences at Johns Hopkins University School of Medicine, spatial biology research in cancer must trend beyond mapping the cellular composition of the tumor microenvironment to understanding mechanisms of immunotherapy response and resistance. In other words, how are individual tumors responding to therapy, where are they going next, and how does this guide therapeutic decisions?
To do that, she and her team have been using Visium spatial transcriptomics to study patient tumor samples from responders and non-responders to a combination immunotherapy against liver cancer. The patients were enrolled in a 2021 clinical trial that tested a combination of cabozantinib (a small molecule inhibitor of tyrosine kinases) and nivolumab (a checkpoint inhibitor), and 5 out of 15 showed a major or complete response. Dr. Fertig’s goal in using spatial transcriptomics on patient samples was to understand the heterogeneity of the tumor-immune microenvironment and determine how this might be influencing therapeutic outcomes.
She and her team found one sample from a responder to be particularly interesting: spatial clustering indicated there were two distinct tumor lobes in the same tissue section, with one lobe showing clear immune infiltration and the other little infiltration. Rather, it was densely packed with fibroblasts. This finding led the team to go back to their physician collaborator on the project, who hypothesized this region might be an indicator of resistance. In the cohort of responders, this patient was the only one to later recur.
“For me, as a computational biologist, this was a very powerful moment of working in human samples and really thinking about the impact of what we’re studying.” – Elana Fertig, PhD
To deeply understand this patient’s immune response and define pathways potentially associated with resistance, Dr. Fertig’s team began looking at patterns of gene expression resulting from tumor and immune cell interactions using a tool they developed, called SpaceMarkers (2), which identifies contributing gene expression pathways from regions of the tissue where tumor and immune cells are overlapping (Figure 1). Their approach further refined the spatial patterning of pathways associated with successful immune-killing and pathways involved in fibroblast-mediated blocking of the immune response.
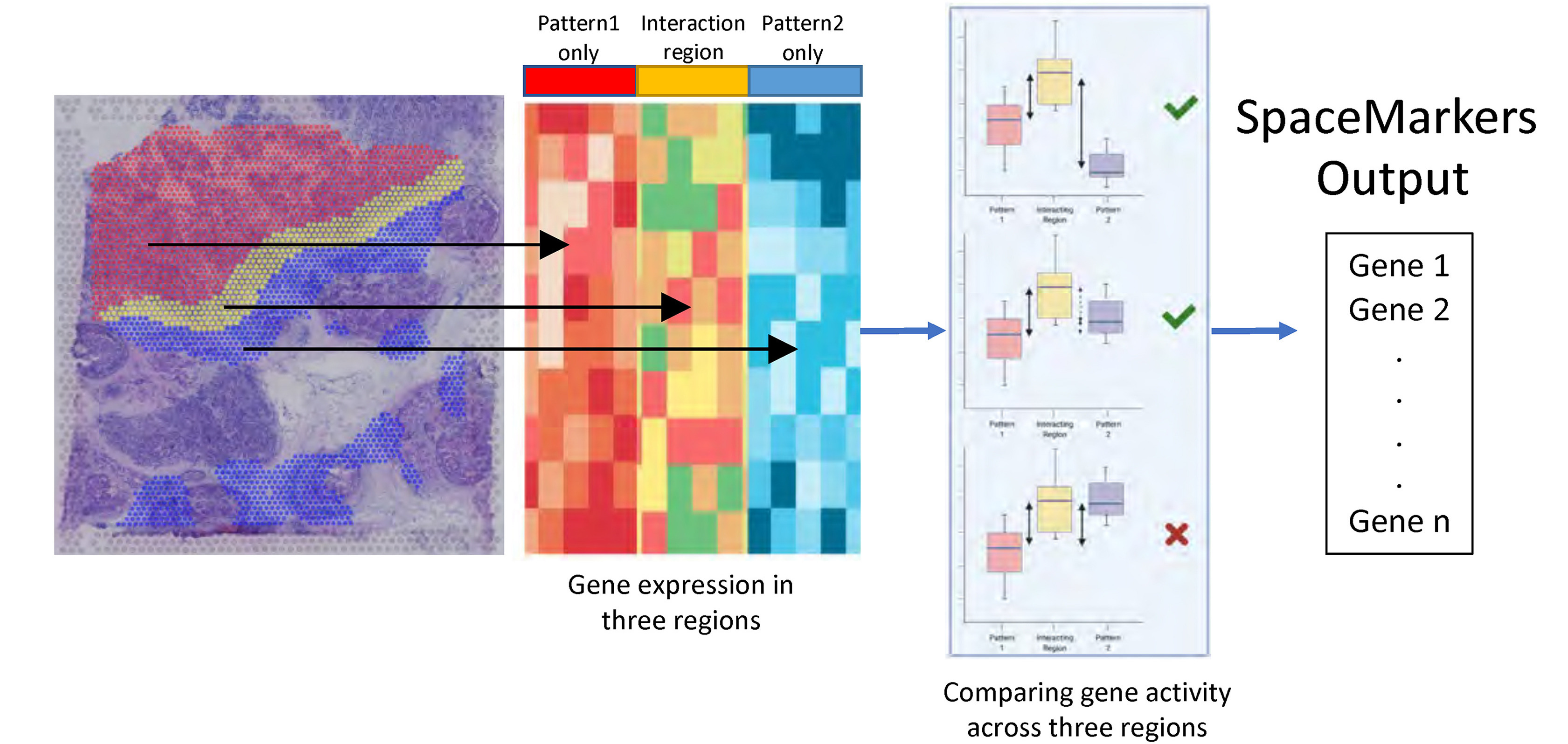
With access to these deeper patterns of spatial gene expression, Dr. Fertig advocated for the application of mathematical models to take their findings to the next step—to project how tumors will respond over time. “If we know the laws of the system, we can write down the equations that describe them and very clearly write out where they’re going in the future.”
This innovative shift in thinking from a data-driven paradigm to a mechanistic mathematical paradigm has led Dr. Fertig and her collaborators to start modeling virtual clinical trials, applying therapies to virtual tumor models seeded randomly with different groups of immune and tumor cells, and then tracking how the system changes over time, including if the tumor is therapy-responsive.
“How do I know if this is real? How do I know if it reflects real tumor biology?” Dr. Fertig argued that this is exactly where spatial transcriptomics data can play a crucial part in cross-comparing and calibrating models with real biology. She conjectured that perhaps spatial transcriptomics data can accurately capture the end-point of a therapy in human spatial molecular data obtained through surgery, and then their simulations can work backwards temporally to understand the starting point and differentiate tumor characteristics between responders and non-responders prior to treatment. This could be a method to identify predictive biomarkers for positive clinical response or resistance, such as cancer stem cells signatures, which they identified through their simulations based on the end-point sample from the liver cancer patient who later recurred.
According to Dr. Fertig, this forecasting approach based on spatial transcriptomics data could take the field closer to precision medicine; but it could also reshape how we think about cancer, more fundamentally, as a chronic illness that changes over time, rather than a static diagnosis. If researchers and doctors can anticipate those changes, perhaps they can better intervene with therapy across the stages of disease.
Back to top2D spatial analysis and 3D modeling meet in the tumor–immune contexture of melanoma
Peter Sorger, Professor of Systems Biology at Harvard Medical School, made the AACR 2024 audience grapple with the limitations of two-dimensional spatial analysis in light of the true complexity of the three-dimensional tumor microenvironment, chalk full of densely packed cells and networks of cellular interactions driving disease or influencing therapeutic response. He noted how a lack of visibility into a cell’s complete three-dimensional composition could potentially lead to incorrect phenotyping, if, for example, transcripts are polarized within the cell, but cut off as a result of thin tissue sectioning; and how a limited view of a cell’s overlapping neighbors (including the cells above it) could lead to misassigned transcripts.
His team’s solution? An immunofluorescence and microscopy-based platform called cyclic immunofluorescence (CyCIF) that maps protein markers to cells within a 35-µm thick tumor section; then, with a commercially available AI microscopy image analysis software called Imaris, visual reconstruction and modeling of the “3D” tumor microenvironment and the heterogeneous tumor–immune interactions within it. Using this approach, Dr. Sorger demonstrated a striking visualization of the interactions between T cells, a dendritic cell, and a tumor cell within an immune community in a melanoma section.
Where do these models meet, and potentially complement, two-dimensional spatial transcriptomics solutions? Access to spatial gene expression within the tumor microenvironment is an essential readout of the cellular composition of the tissue system, as well as the functional relationship between spatial localization in the tumor microenvironment, which is scattered with vasculature, necrotic regions, and inflammatory niches, and activated cell type–specific gene expression programs. With that in mind, Dr. Sorger commented on the value of visualizing the genotype–phenotype relationship in cells by mapping back and forth between a gene expression readout, aided by Xenium In Situ single cell spatial imaging, and a morphology readout, aided by his proteomics and 3D modeling approach. His team leveraged Xenium with a 500-plex panel to study a metastatic melanoma sample, demonstrating concordance with the CyCIF approach to identify NGFR+ tumor cells; then, with the additional morphological readout, zoomed in further to deeply phenotype individual cells, including a strange multinucleated tumor cell.
Dr. Sorger anticipates further innovation of machine learning algorithms that can pull insights from these 2D spatial gene expression and tissue imaging analyses, which can be scaled to many samples, and 3D reconstruction approaches to ultimately optimize single cell phenotyping.
Back to topInvestigating tumor clonality in breast and prostate cancer with infCNV and high-definition spatial transcriptomics
Patient tumor samples are rare and precious, making it essential that researchers maximize the information they can obtain from a single tissue section. With that need in mind, Joakim Lundeberg, PhD, a group leader at SciLifeLab in Stockholm, Sweden, and head of the division of Gene Technology at KTH Royal Institute of Technology, and his team have been optimizing spatial multiomics approaches through applications of Visium whole transcriptome spatial gene expression data and bioinformatics tools.
Dr. Lundeberg first established the value of a whole transcriptome spatial readout in exploring tumor ecosystems and deriving biological insights that morphological analysis alone cannot reveal. This includes mapping the presence of cellular niches that pathological annotations may not identify (including notorious tertiary lymphoid structures (TLS), which Dr. Lundeberg noted can be a predictive biomarker of immunotherapy response and can be scored and identified via a mix of B- and T-cell transcripts from spatial data). Moreover, spatial gene expression—with the additional enhancements provided by deconvolution methods using patient-matched single cell datasets—can help researchers characterize cell types in morphological context, discover cellular colocalization and interactions, and distinguish the biology of the tumor core from the tumor edge.
But there’s more that can be seen from this readout. Dr. Lundeberg’s research group has been applying a computational method (infCNV analysis) that infers copy number variation (CNV) information from gene expression data, spatially resolving genetic gain or loss events that could be underlying tumor clonal evolution and cancer progression. Using this method in a human prostate cancer sample, their team defined a spatial clonal tree demonstrating genetic events along the axis from benign epithelial cells, to an altered benign epithelial cell state (which was previously annotated as normal epithelial by morphological analysis), to three distinct tumor clones. The ability to perform spatial transcriptomics in FFPE human prostate tumor sections allowed them to explore additional archived samples to confirm these genetic events and demonstrated the presence of altered benign epithelia as well as tumor clones overlaid onto specific gland structures in the tumor microenvironment. Importantly, tumor clone profiles can be traced in spatial transcriptomics data from primary tumor samples and patient-matched metastasis tissues, such as in the lymph node, which may help researchers understand tumor evolution and the clones driving metastasis.
Spatial resolution is the key to unlocking even deeper insights, whether resolving tumor clones or tracing TLS regions and the cell types within them. Thus, Visium HD has helped Dr. Lundeberg’s research group take their studies to the next level. Commenting on the 2 µm barcoded squares arrayed in the Visium HD Capture Area and data binning strategy, Dr. Lundeberg said,
“If you bin [the information from 2 µm] into 8 µm, 16 µm, 48 µm, you can actually start to see where your cell types are by deconvolution. Now we have a way to explore transcriptional patterns at the single cell resolution. And the binning is quite helpful, because you can look at single cells and cell states at the 8 µm [level]; at 16 µm, you can look at pairs of cells which are interacting; or you could go up to 48 µm, and then you can see clusters of cells like the TLS structures. This is really useful because you can gear your questions to the different resolutions.”
Looking at Visium HD data on human triple-negative breast cancer samples, which their team generated just two days before AACR 2024, Dr. Lundeberg showed the value of aligning insights from histology, spatial gene expression, and inferred CNV events. A pathologically annotated in situ cancer region had marked upregulation of gene expression signatures associated with TLS immune aggregation; in that same region, infCNV identified a unique tumor clone, segregated from 4 other clones present globally in the section (including an invasive region). With these high-resolution insights, the team could start to associate CNV events with specific cell types present regionally and further define the B- and T-cell states contributing to the TLS signature.
We’re incredibly excited to see how Dr. Lundeberg’s group and others continue to go deep into cancer biology with whole transcriptome spatial gene expression and innovative multiomic techniques at, now, single cell–scale spatial resolution with Visium HD.
Back to topSingle cell and spatial FFPE analysis makes more comprehensive study of premalignant lesions for lung adenocarcinoma possible
Lung adenocarcinoma (LUAD) presents a particular challenge to the cancer community because of major gaps in our ability to identify patients at high risk for disease. In her talk at AACR 2024, Ansam Sinjab, PhD, Research Scientist at MD Anderson Cancer Center, introduced a factor that has stalled progress in early detection for this cancer type—particularly, an inability to access and deeply characterize an earlier stage of LUAD called adenomatous premalignant lesions (aPMLs) from human patient samples. These lesions are small and rarely surgically resected, but if they are, they’re preserved by formalin fixation and paraffin embedding (FFPE), making them difficult to analyze with high-resolution single cell and spatial tools.
That is, until recent advances that have made single cell RNA-sequencing and spatial transcriptomics experiments possible in FFPE tissues. With these matured technologies, Dr. Sinjab sought to define the mechanisms underlying pathogenesis, including the molecular profiles, cell states, and cell–cell interactions that evolve along the progression axis from normal lung to the premalignant state to LUAD.
Leveraging archived human FFPE samples with matched LUADs, aPMLs, and lesion-adjacent normal-appearing tissues (NATs), which are at risk to progress to LUAD, Dr. Sinjab applied a suite of FFPE-compatible tools to study the underlying biology of cancer progression, including single cell RNA-sequencing (enabled by Chromium Gene Expression Flex), spatial transcriptomics with protein co-detection enabled by Visium, high-plex immunofluorescence, and, in some samples, subcellular spatial transcriptomics with Xenium single cell spatial imaging. Integrating data from these platforms could be the key to revealing rare cellular subsets driving progression to LUAD and identifying biomarkers associated with a high risk of disease.
This multimodal analysis was further guided by her group’s recent single cell findings that suggest high-keratin-expressing KRT8 alveolar cells (KACs) may serve as LUAD intermediates, which was also informed by spatial transcriptomics data that localized KACs to tumor regions and lesion-adjacent regions. Spatial transcriptomics data of their matched aPML and LUAD samples demonstrated an increase in KAC gene expression signatures in the course of tumor evolution. They also found, in immunofluorescence and Xenium data, that KACs interacted with CD68+ macrophage cells within the normal-appearing lesion-adjacent tissue. Visium spatial transcriptomics data then helped to identify that interleukin 1-beta signaling, which is a key mediator of the inflammatory response, is high in macrophages within this tissue compartment, pointing to a possible mechanism by which KACs emerge in premalignant tissues.
Dr. Sinjab’s multimodal analysis revealed different immune cell characteristics that may also play a role in cancer progression. Single cell analysis demonstrated a significant increase in regulatory T (Treg) cells in LUAD samples; Visium spatial transcriptomics data showed a few scattered clusters of the Treg signature in aPML tissues, but a highly concentrated Treg signature near tumor cells in the matching LUAD samples. Xenium data indicated B cells were aggregating to tertiary lymphoid structures—known sites of persistent inflammatory stimulation—in the tumor tissue, while Visium data further demonstrated B cells increased in proportion and complexity within tertiary lymphoid structures between matched premalignant and LUAD samples. These findings may point to a progressive, immune-mediated mechanism of cancer development that could be further explored to identify targets for the early stages of disease.
Decoding the tumor ecosystem at multiple stages of disease progression, from multiomic angles, and with multiple high-resolution technologies could be a game-changing approach to, ultimately, trace all of the complex biology that contributes to cancer—and provide actionable insights to improve detection and treatment in the future.
Back to topWorking smarter and harder against cancer
The studies that we’ve highlighted in this blog each represent innovative research from incredibly talented scientists who leveraged single cell, spatial, and computational toolkits to ask deeper questions about cancer—to work harder and smarter against this complicated disease.
But, more importantly, these studies represent a step forward in understanding and, eventually, treating cancers that affect millions globally: breast cancer, liver cancer, melanoma, lung adenocarcinoma. It’s a humbling and sobering responsibility to connect these tools to the brave researchers that will go on to influence clinical outcomes in cancer. It’s the possibility of actually improving medicine and patient outcomes that keeps us eager to meet you, the cancer community, at the edge you’re leading in technology and computational development, and to continue working to enable your next moonshot.
We wish we could’ve highlighted all of the amazing studies that used 10x Genomics technology at AACR 2024! To continue exploring where we were at the conference, please find our AACR 2024 posters and other literature here.

If you’re interested in learning more about the 10x Genomics technologies highlighted in these studies, please explore some of our other resources:
-
Xenium In Situ (imaging-based, targeted spatial transcriptomics at single cell resolution)
-
Visium HD (sequencing-based, whole transcriptome spatial gene expression at 2-µm resolution)
-
Chromium Single Cell Gene Expression
- Chromium Gene Expression Flex (probe-based single cell gene expression for FFPE tissue, in addition to other tissue preservation conditions)
- Chromium Single Cell Gene Expression v4 (3’ RT-based, now powered by GEM-X technology)
- Chromium Single Cell Immune Profiling v3 (5’ RT-based, now powered by GEM-X technology)
References:
- Kumar T, et al. A spatially resolved single-cell genomic atlas of the adult human breast. Nature 620:181–191 (2023). doi: 10.1038/s41586-023-06252-9
- Deshpande A, et al. Uncovering the spatial landscape of molecular interactions within the tumor microenvironment through latent spaces. Cell Syst 14: 285–301.e4 (2023). doi: 10.1016/j.cels.2023.03.004
About the author:
