This page summarizes key changes to the new web summary layout to locate data quality control (QC) components.
The cellranger multi pipeline generates a web_summary.html file, which provides an overview of the analysis performed on the data and serves as a starting point for quality control. This file includes key summary metrics, such as sequencing depth, cell counts, and data quality indicators, presented as tables and visualizations for easier interpretation.
The interactive format allows users to explore the results with hover-over tooltips and clickable tabs, making it convenient to review and share important insights without needing separate analysis tools. If an issue is detected during the pipeline run, an alert appears on this page (see the Cell Ranger troubleshooting documentation for details).
For assistance with understanding metrics or charts, see the help text within the web summary HTML. The help text for tables and charts in the web summary UI has been improved to provide clearer guidance on interpreting key metrics, understanding their calculations, and using them to assess data quality.
The metric and plot definitions have not changed, so existing documentation about web summary metrics still applies. Here is a list of available web summary Technical Notes:
- Interpreting Cell Ranger Web Summary Files for Single Cell Gene Expression Assays
- Interpreting Cell Ranger Multi Web Summary Files for Single Cell 3’ Gene Expression with Feature Barcode technology for Cell Multiplexing
- Interpreting Cell Ranger Web Summary Files for Chromium Single Cell Immune Profiling
- Interpreting Cell Ranger multi Web Summary Files for Flex
In this section, we will guide you through using the multi web summary to perform QC on your experiment. Although this example focuses on a multiplexed Flex Gene Expression and Antibody Capture experiment, most of the QC metrics and charts shown here are applicable to other library combinations, and the QC workflow interpretation applies to all web summaries.
Web summary tabs
Your web summary includes multiple tabs based on your experimental design, such as the number of samples, inclusion of Feature Barcode libraries, and cell type annotations. The first tab contains metrics specific to the Gene Expression library and serves as the default tab for any experiment containing a Gene Expression library. This tab is an ideal starting point for QC analysis, offering essential sequencing and mapping metrics for evaluating library quality.
If your experiment includes a Feature Barcode library, such as Antibody Capture, CRISPR Guide Capture, or Antigen Capture, the results for each library will appear in a separate tab named accordingly (one tab per included Feature Barcode library). In this example, an "Antibody" tab displays the results specific to the Antibody Capture library.
If you multiplexed samples using one of 10x Genomics' multiplexing solutions (such as on-chip multiplexing, hashing with Antibody Capture, or 3' CellPlex), an additional tab labeled Samples will provide sample-level metrics. Each sample in your experiment will have its own web summary, and examining the sample-level metrics for each can help identify any outliers that may impact overall experimental quality. If you performed cell or sample hashing with Antibody Capture, both an Antibody tab and a Samples tab will be present.
If cell type annotations were enabled in your multi run, an additional tab will appear, displaying the annotation results.
Finally, all multi web summaries include an Experimental Design tab, which provides a visualization of the experimental design as inferred by Cell Ranger based on the multi config CSV file you provided.
Begin your QC analysis with the library-level metrics, as these sequencing and mapping metrics are foundational for assessing library quality. If these metrics show issues, it likely indicates compromised quality across the library and potentially poor sample-level metrics (multiplexed experiments). First, verify the number of barcodes assigned as cells (cell calling). Start by checking the estimated number of cells metric to ensure that it aligns with your expectations.
Use the barcode rank plot to confirm the expected number of cell-associated barcodes and to check for single cell behavior. An unusual shape in the barcode rank plot at the library level may suggest issues with GEM well loading or a potential wetting failure. Refer to the sample-level barcode rank plot to confirm single cell behavior for this sample (described in the sample tab).
Next, look at the fraction of reads in cells metric. A low fraction of reads in cells confirms the absence of single cell behavior. Since this is a library-level metric, check all the samples in your experiment to identify any problematic sample. Excluding problematic samples from downstream analysis may improve results.
Lastly, the fraction of initial cell barcodes passing high occupancy GEM filtering metric helps verify proper chip loading. A high value here indicates successful chip loading, while lower values suggest data loss, which may require cautious downstream analysis.
Use the mapping metrics to assess whether your reads mapped well to the reference. Each metric in the mapping quality section provides insights into mapping quality.
-
Reads mapped to the probe set: Lower values may indicate low total expression, use of incorrect probe set, suboptimal sample preparation, or the use of input FASTQs from the wrong assays (i.e., non-Flex).
-
Reads confidently mapped to the probe set: This fraction should also be high. If reads mapped to the probe set are high but reads confidently mapped are low, run FastQC to troubleshoot.
-
Reads half-mapped to probe set and reads split-mapped to probe set: Evaluate these metrics to check for partial mappings. Higher values for these metrics may result from low RNA content in the sample, inadequate washing after probe hybridization, deviations from the recommended protocol during hybridization, or suboptimal sample preparation.
Check the sequencing depth to ensure sufficient total reads and reads per cell. If these metrics look satisfactory, proceed to the following metrics to confirm read quality:
- Valid Barcodes
- Valid UMIs
- Valid Probe Barcodes
- Valid GEM barcodes
If you observe low values for the above four metrics, check the Q30 metrics and consult your sequencing provider for further guidance.
For Flex experiments, this section helps assess whether the distribution of UMIs and cells among samples is as expected.
-
UMIs per probe barcode: Check the number and fraction of UMIs for each Probe Barcode among all UMIs for that library type. The fraction of the UMIs should be similar between probe barcodes if the associated samples are expected to have similar complexity and target cell numbers.
-
Cells per probe barcode: Similarly, the number and fraction of cells per Probe Barcode among all cells detected in the GEM well should be similar if the associated samples are expected to have similar complexity and target cell numbers.
Will sequencing deeper help?
Use the sequencing saturation plot and median genes per cell plot to assess if increased sequencing depth could improve your results. If the slope of the median genes per cell curve near the endpoint is flat, it suggests saturation, indicating that more sequencing depth is unlikely to increase transcript complexity.
Is there genomic DNA noise?
The estimated UMIs from genomic DNA metric should be less than 1% under optimal experimental conditions. Similarly, the estimated UMIs from genomic DNA per unspliced probe metric should be low.
Evaluating transcript complexity
For each sample, examine the median genes per cell plot on its web summary to check if there is sufficient transcript complexity. The curve's slope near the endpoint offers an estimate of the potential gain from increasing sequencing depth. A flat slope indicates saturation, suggesting that additional sequencing will not enhance transcript complexity.
If you observe distinct structures or unexpected patterns in these visualizations, contact 10x Genomics Technical Support for further troubleshooting or filter the data and proceed with caution.
Checking expression of specific genes
To see if specific genes of interest are highly expressed in your cells:
- Examine the t-SNE projection of cells colored by cluster to identify clusters of interest.
- Review the Top Features By Cluster table for key gene expression patterns.
The Antibody Library tab contains metrics and visualizations at the library level for the Antibody Capture library. If there is a problem with your Antibody Capture library, an alert will appear at the top of this page.
Start QC for this library by ensuring that the correct Feature Barcode reference was input into the multi config CSV by looking at the Feature Reference section under that tab name.
The Cells count reported in this section matches the library-level cell calls. Check the Fraction antibody reads to confirm that this value is high. If the percentage is low (less than 70%), verify that the sections in your Feature Reference CSV were correctly specified. Common issues include incorrect entries in the sequence or pattern fields. Additionally, low-quality reads may contribute to a lower fraction of antibody reads.
The Antibody reads in cells section reports the fraction of valid-barcode, valid-UMI, recognized antibody Feature Barcode reads associated with cell barcodes. This metric should also be high. Low values can result from various factors, including:
- Poor cell washing
- Suboptimal sample quality
- Emulsion failures
- Suboptimal antibody titration and labeling methods
- Low target population (e.g., use of rare markers)
- Low sequencing depth, which can impact cell-calling algorithms
The Fraction of antibody reads usable should ideally exceed 20%. This metric can be affected by a low number of antibody reads in cells and a low fraction of antibody reads.
The Fraction of antibody reads in aggregate barcodes metric shows the fraction of read pairs with valid barcodes that were removed due to aggregation. Ideally, this value should be below 5%. Higher values may result from poor sample quality, inadequate washes, or suboptimal staining and antibody titration. Pre-spinning antibodies is also essential to remove protein aggregates.
Evaluate the sequencing quality of the Antibody Capture library in a similar way to the Gene Expression library. A key metric to examine is Mean reads per cell, which is calculated as the total number of sequenced antibody read pairs divided by the number of cell-associated barcodes.
The Metrics per probe barcode section can also be evaluated similarly to the Gene Expression section. The UMIs per probe barcode column reflects UMIs specifically from antibody reads. The Cells per probe barcode column pertains to the entire Gene Expression library and should match the values reported in the Gene Expression library tab.
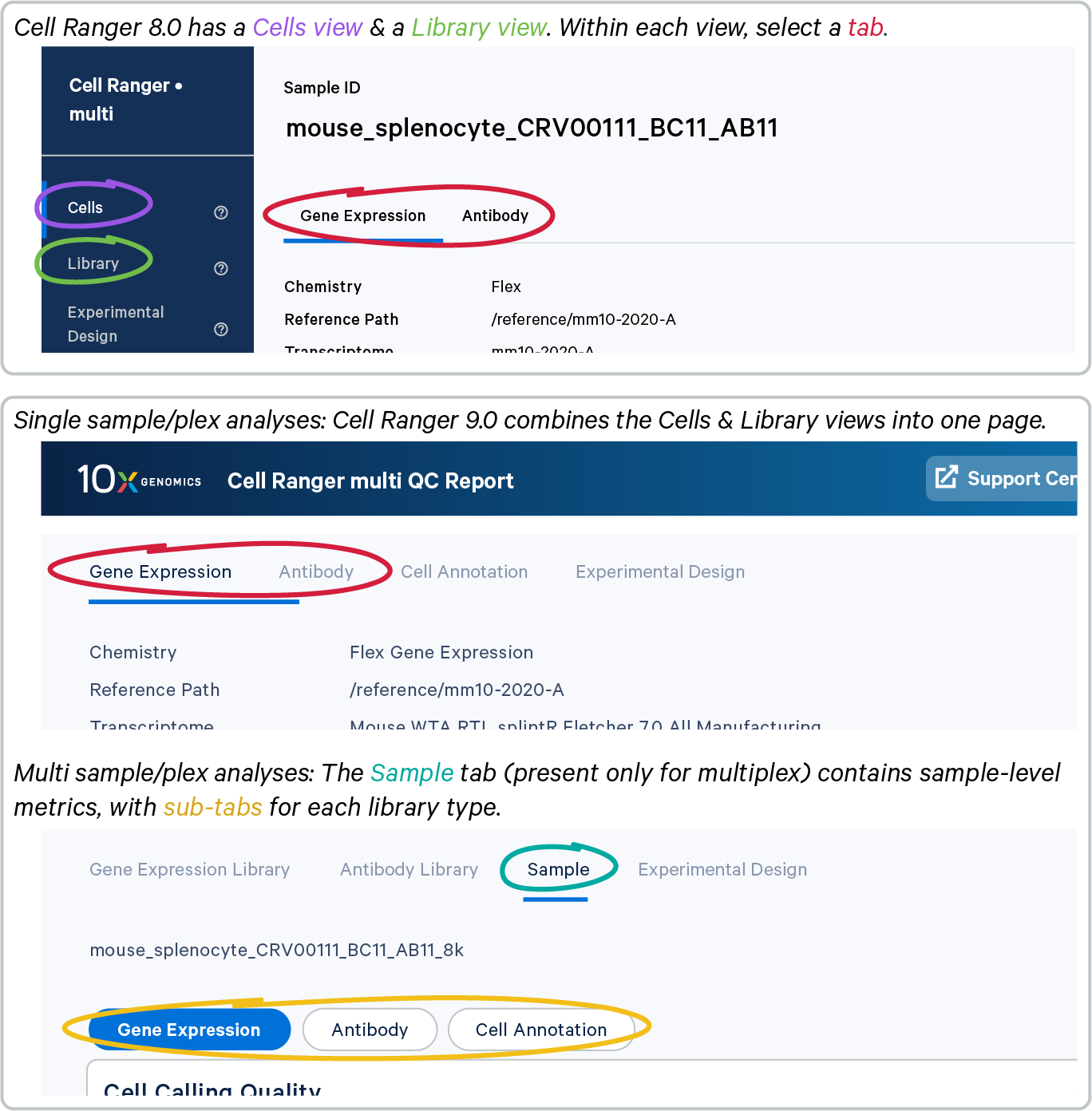
On the Sample tab, double-check the sample ID. In a multiplex experiment, there will be one web summary per sample. Sample tabs are not present for singleplex experiments.
The Sample tab includes one or more subtabs based on your experimental design, such as Gene Expression, Antibody (or other Feature Barcode library), and Cell Annotation (if cell annotations were enabled in the multi run).
The Gene Expression tab contains the most important information for evaluating sample quality. The order of subsections for cell calling and mapping metrics is identical to those presented in the Gene Expression Library tab. However, all metrics and visualizations in this tab pertain specifically to this particular sample.
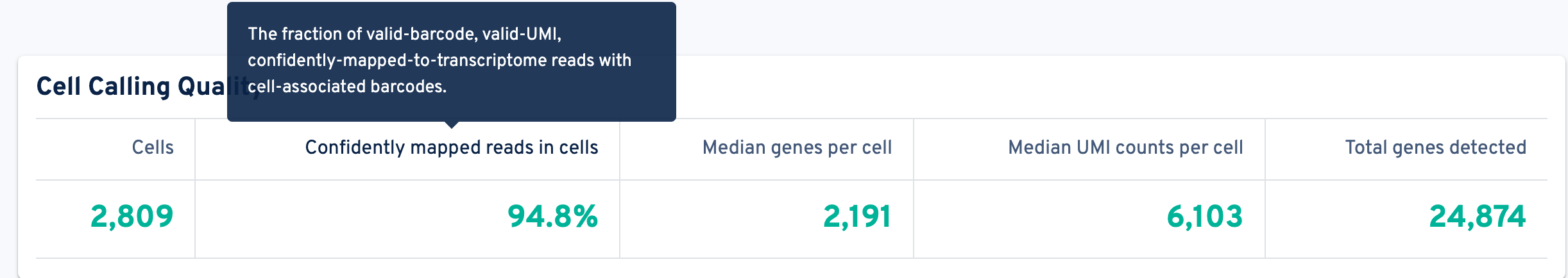
Start by evaluating the metrics in the Cell Calling Quality section. The cells section contains the number of cells called for this sample. Higher or lower than expected values may indicate inaccurate cell count, large amount of debris, poor sample quality with extremely low RNA content, or failures during GEM generation.
Use the barcode rank plot to confirm the expected number of cell-associated barcodes and to check for single cell behavior. Single cell behavior is typically indicated by a steep drop-off in the graph (an elbow-knee shape) that clearly separates cell barcodes from background barcodes (starting at the knee). Refer to 10x Genomics' Interpreting Cell Ranger multi Web Summary Files for Flex Technical Note for help interpreting the sample-level barcode rank plot.
If you observe over- or undercalling of cells, consider rerunning the analysis with the --force-cells option set to the expected cell number. For overcalling issues, you can also use Loupe Browser to add filters and recluster based on your experimental criteria.
-
Median Genes per Cell: This metric is calculated based only on reads associated with "cells" in the particular sample. Do not compare this metric to the sequencing recommendation (10,000 mean reads per cell), as the recommended sequencing depth is calculated at the library level (mean reads per cell), not the per-sample level. The required number of reads per cell for each sample depends on the cell type (high or low RNA content) and the desired analysis.
-
Median UMI Counts per Cell and Total Genes Detected: These metrics should also be high. Lower than expected values may indicate shallow sequencing depth and/or compromised sample or library quality.
The Mapping Quality section provides the same information as in the Gene Expression Library tab, but specifically for reads from cells assigned to this sample. Examine this section carefully for each sample in your experiment to identify any outliers that may be biasing data quality.
The UMAP projections are shown for this sample's cell barcodes, with each cell color-coded according to its UMI count. Cells with higher UMI counts likely have higher RNA content than those with fewer UMIs. The Interpreting Cell Ranger multi Web Summary Files for Fixed RNA Profiling Technical Note provides guidance on interpreting clustering patterns. Although the descriptions are for t-SNE, they also apply to UMAP.
Use the Top Features by Cluster (Log2 fold-change, p-value) list to check if it is dominated by mitochondrial genes. While some samples naturally exhibit high mitochondrial gene expression, most samples with predominantly high mitochondrial gene expression often have a large number of dead or dying cells, indicating poor quality.
The Antibody Expression metrics are presented for reads in this sample. Interpretation is similar to the Antibody Library tab.
The Histogram of Antibody Counts helps explore gene expression levels of the top 120 antibodies (by UMI count) across the library's cell population. Only antibodies with total UMI counts over 1,000 are plotted. The X-axis represents UMI counts on a log scale, while the Y-axis shows the number of cells with that UMI count. To view this histogram for specific antibodies, select (or deselect) antibodies by clicking on the legend labels.
The Antibody tab includes a Distribution of Antibody Counts plot, which displays the relative composition of antibody counts for antibodies with at least one UMI. The box size is proportional to the fraction of total UMIs from cell barcodes associated with that antibody. Hovering over a box provides additional metrics and information.
If you ran the cellranger multi pipeline with cell annotations enabled, a new Cell Annotations subtab appears under the Sample tab. This tab provides key statistics and visualizations for exploring cell types within your sample. For detailed descriptions of these metrics and visualizations, see the cellranger annotate page.
The multi web summary has been redesigned to simplify navigation and improve clarity. Previously, the summary was divided into three views: Cells, Library, and Experimental Design. To streamline the user experience, these distinct views have been removed and consolidated as follows:
- Cell and library information:
- Singleplex: The contents from the Cells and Library views are now combined into a single tab, bringing related metrics together for easier review.
- Multiplex: The Gene Expression Library tab displays library-level metrics by default, while sample-level metrics are available in the Sample tab.
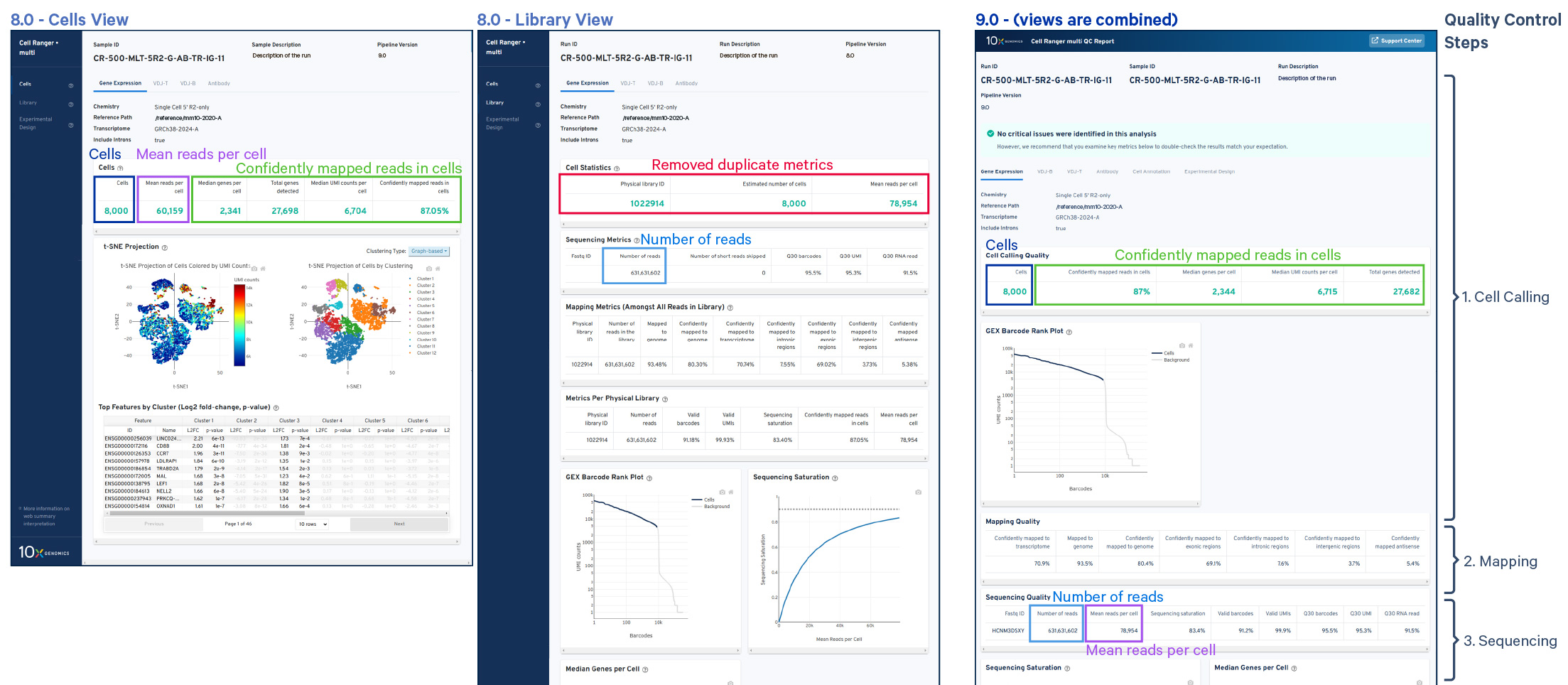
- Improved metric organization: Metrics have been reorganized, grouping metrics that are frequently reviewed together within the same table. Some metrics have also been moved within the QC workflow for improved relevance and logical flow (see image).
-
Removed redundant and uninformative metrics: Duplicate metrics have been removed, and a few metrics deemed uninformative have been excluded from the summary to reduce noise and focus on the most critical data.
-
Enhanced tooltip help text: Hovering over metrics now displays help text, offering quick, accessible information about each metric without cluttering the page.