Getting started with Visium HD data analysis and third-party tools

Visium HD is a spatial biology discovery tool that generates whole transcriptome data at single cell scale from FFPE, fresh frozen, and fixed frozen human and mouse tissues. It uses NGS-based spatial transcriptomics to map the molecular expression profiles of diverse cell types while preserving the complex spatial networks within tissue architecture. Software tools like Space Ranger and Loupe Browser, available for free with Visium HD, unlock the insights from this unique data modality which combines a histological image with high-resolution spatial RNA sequencing data. Easy integration with a growing ecosystem of R- and Python-based community developed tools that enable cell segmentation, cell type deconvolution, and spatial neighborhood analysis, among other capabilities, promises even greater discovery potential.
In our recent webinar, we provided an overview of the Space Ranger pipeline—now also supported with 10x Genomics Cloud Analysis—and some of these community tools. As a follow-up, we wanted to condense the expertise of our computational biology team into a Q&A that helps provide a starting point for you to learn more about Visium HD data analysis and some tips for how to approach third-party tools.
Keep reading to review our conversation with 10x Genomics Staff Computational Biologists, Stephen Williams, PhD, and Joey Arthur, PhD, who helped to develop the Visium HD software solutions. Learn how to get started with Visium HD data analysis using Space Ranger and Loupe Browser, and find recommendations for third-party analysis tools that enable integration of single cell data with Visium HD data, cell segmentation, and more.
How to get started with Visium HD data analysis
This interview was conducted with Stephen Williams, PhD, Staff Computational Biologist at 10x Genomics. It was edited for length and clarity.
Could you describe the recommended path to analyze Visium HD data? How would someone go from the end of their experiment, the sequencing step, to navigating their HD data?
Stephen: The very first thing that you do after you get your sequencing data would be to run it through the latest version of Space Ranger. That's going to do all your barcode correction, UMI detection, map all of the transcripts to their associated region on the image, and produce all the output files that you're going to need for any third party tools that you'd like to use.
[Our recent webinar provides a simple walk-through of how to run your Visium HD experimental output files through Space Ranger.]
It also produces a Loupe file which is a really good place to start exploring your data. And the cool thing about the Loupe file, especially for Visium HD data, is that your original image is in that file. So it’s super high resolution, or as high resolution as you had on your microscope. You can zoom in and make sure that your alignment looks correct at a very tight scale. You can look at all the genes that you're interested in and do some of that first pass, and even secondary analysis that you'd like to do, like reclustering. Or, for example, where your tumor versus normal boundaries are in your cancer sample.
From there, there's nice support in Seurat, Scanpy, or Squidpy for doing some QC, like say you want to remove bins that would have high mitochondrial content, or you want to set some sort of UMI threshold, or use different clustering parameters.
So that's kind of the first path at your data and the things that I would do generally.
How does someone learn how to do those exact steps? Is there a place they should go to learn how to follow those analysis steps you described?
Stephen: The 10x Genomics support site has every document that you could ever want—like how do you run Space Ranger in great detail, or what image formats we accommodate, which sequencing types you should be using, short read versus long read, and also sequencing configuration. Or, if you do deviate from what we have recommended, you can learn how to enter that into the command line.
It's very easy to install Space Ranger and gives instructions on how to do that. Everything is pre-compiled. All you have to do is download Space Ranger and extract it on your computer or your compute system and it's super easy to get going from there.
Then, as far as that secondary analysis with third party tools—perhaps more high-level filtering and maybe different clustering—they have really good tutorials as well. If you go to the third party tool’s website, they have those tutorials and we actually work closely with those groups to make sure that those tutorials address what we believe would be our users' highest needs.
What should a user expect will be different when they're analyzing Visium HD data versus lower resolution spatial transcriptomics datasets? For example, how would you prepare a customer transitioning from the v1 or v2 Visium Spatial Gene Expression assays to Visium HD?
Stephen: I think the first experience that they'll have is just the striking scale of the data—just how refined things are. We've got images of serial sections done on Visium v2 versus Visium HD. You can tell on v2, this has a nice structure and we can see where the gene expression patterns are different in the tissue. But in HD it's like, wow, this is almost like a pathologist has gone and annotated the tissue because the scale is so fine, even at the 8 µm bin scale. So that's the first thing that people will notice.
The next thing that they will probably notice is this is a lot of data. What do I do with this? I think learning how to use Loupe, which has been optimized to look at the 8 µm scale, should be no problem on your computer. When it comes to the third party tools, we've helped them understand the data and they have adapted their resources to be able to handle such a large volume of gene data generally.
If you were to give a user a homework assignment to get used to Visium HD data, what resource would you send them to? What would you want them to try in that Visium HD data resource?
Stephen: It totally depends. If they are a bioinformatician, I would say go look at the tutorials that Seurat and Scanpy provide—they provide datasets that are preloaded. It's really easy to use. You should be able to go step by step through everything.
If it's a lab person from a lab that doesn't know R or Python, I'd say go straight to that Loupe file, typically at 8 µm. You already know the biology that you're looking for. You understand your tissue. Start typing in the name of your favorite gene and just see where it’s located. Maybe sit down with someone in your lab or a pathologist that knows the structure of the tissue and understands it. There's a lot of information to be gleaned from that and those people are the experts in their field. Understanding your gene expression profiles in the context of the image that you're looking at is one of the best things that they can bring to the table.
We have a lot of datasets from different tissues and species, mouse and human, on our site for people who haven't necessarily done their first Visium HD experiment. I think that you could be easily convinced that this technology is amazing just by going and looking at the public datasets—looking at a web summary, downloading a Loupe file or a matrix and the spatial directories, and just loading it up either in Loupe or your favorite analysis tool. See what it looks like because the visualization of this technology is incredible.
[Want to get started? Explore a step-by-step Visium HD analysis tutorial: Mapping the Tumor Microenvironment with Visium HD and Loupe Browser.]
Are there any cool features of Loupe Browser that you would want to call out for Visium HD analysis?
Stephen: One feature that I really like about Loupe is being able to compare differential gene expression with violin plots between clusters. That gives you a really good idea of how your genes of interest are distributed.
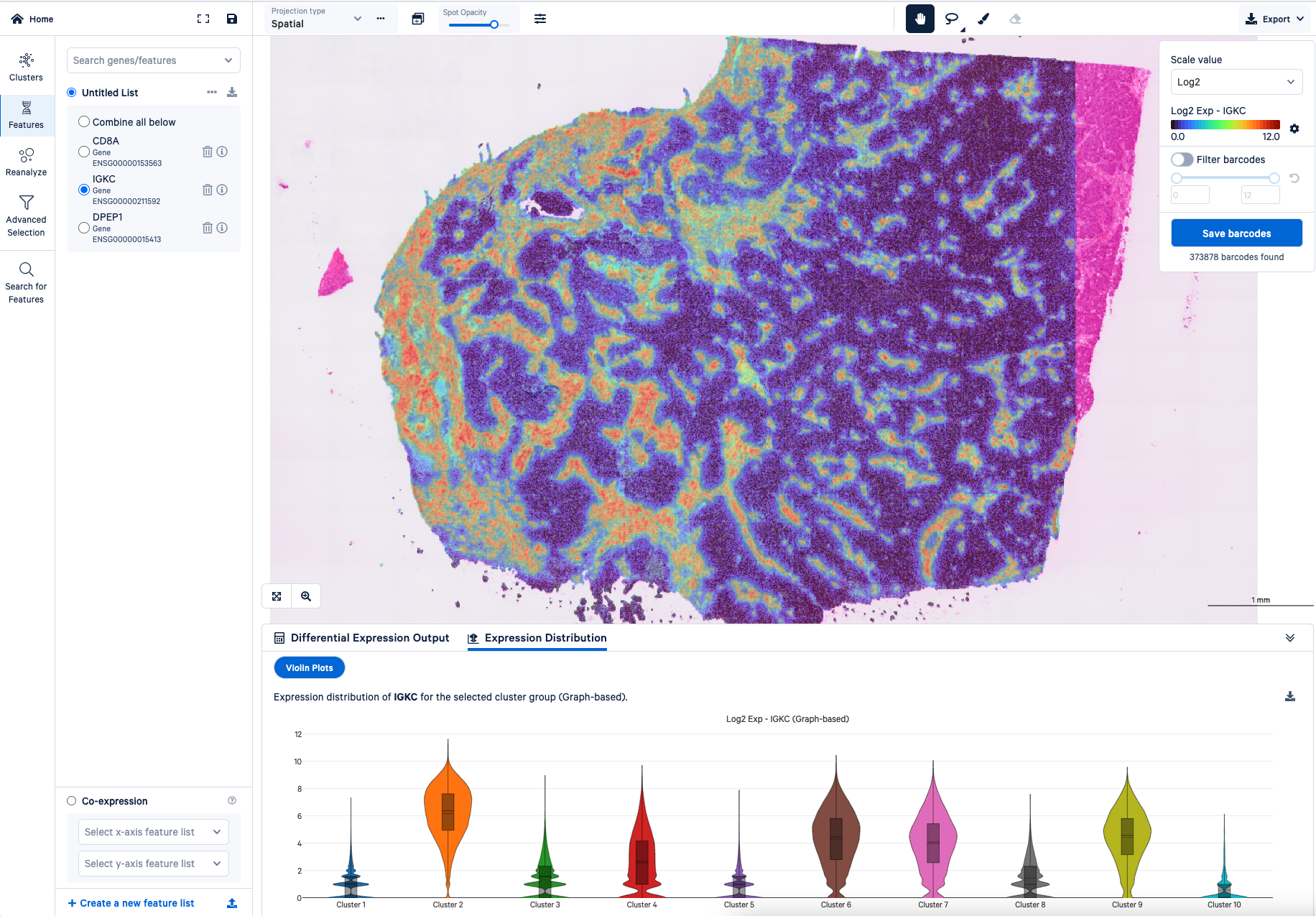
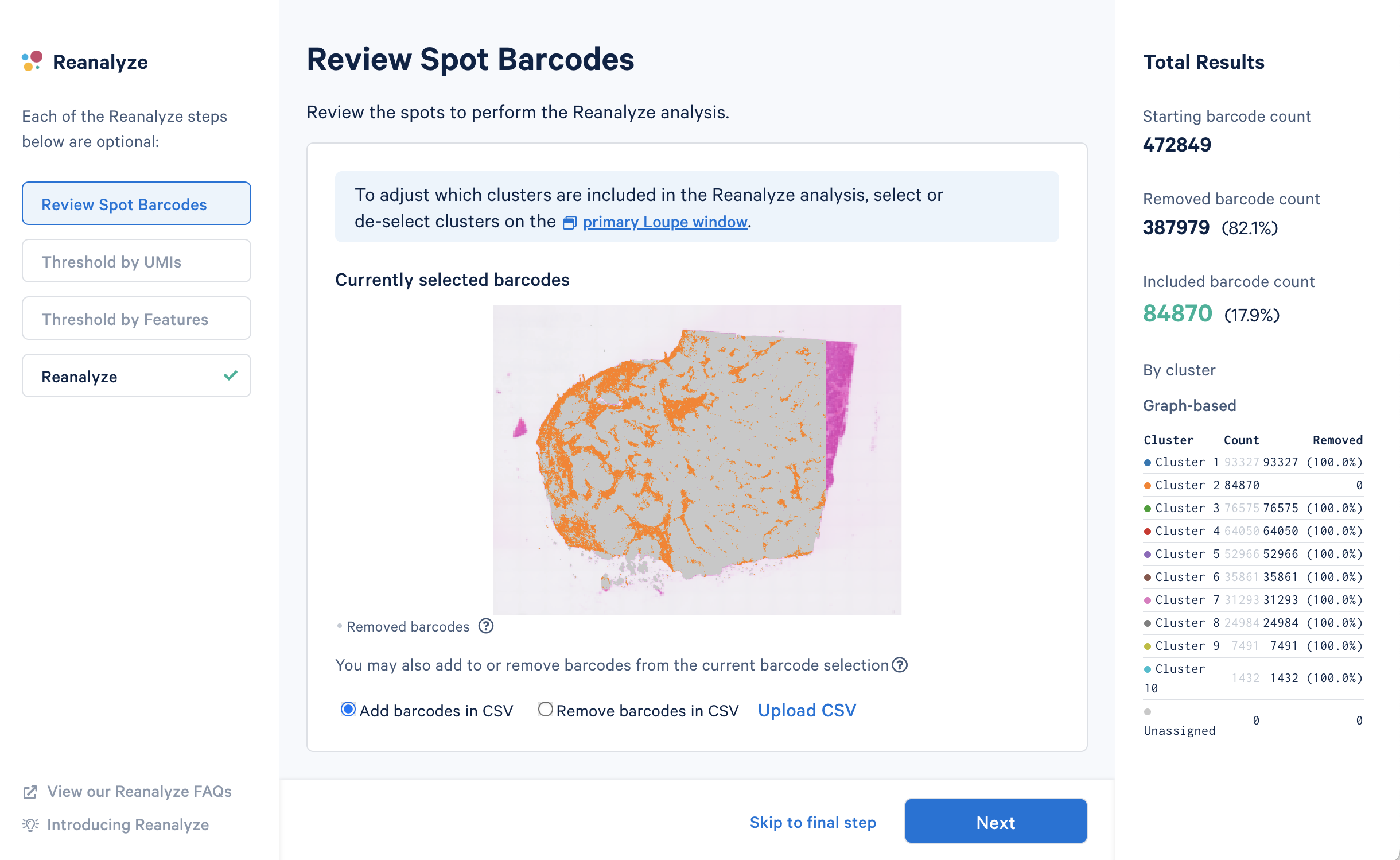
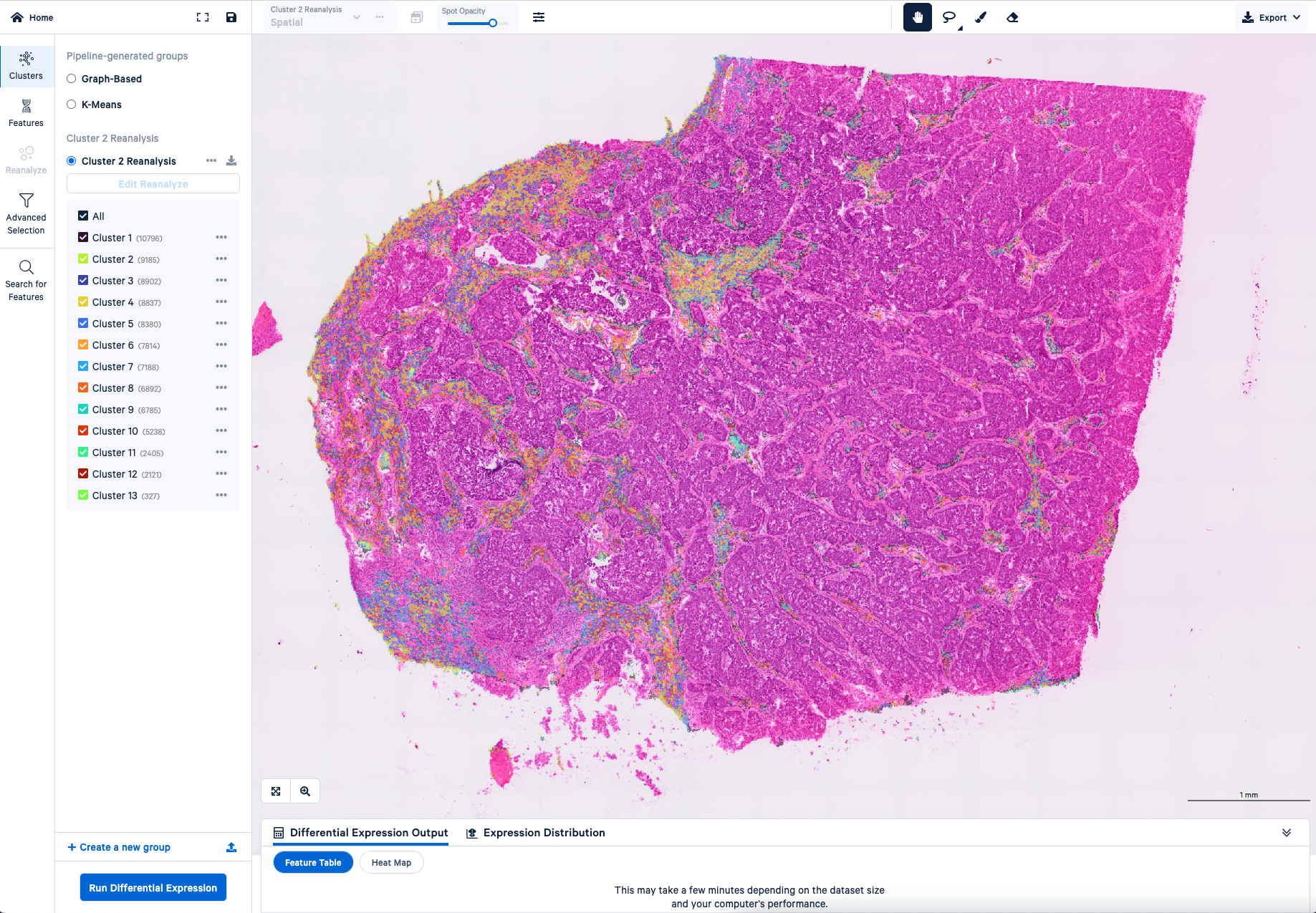
Also, when you know something about your tissue, maybe you think a certain region is under clustered—say in your tumor region—you can select that region and recluster it. Maybe you’ll get a little bit more refined information. That region-specific analysis will work right out of the box, and it's very fast. Without Loupe, if you were to try that same reclustering analysis, you actually need some computer programming skills and it could take a while. That makes Loupe really awesome.
If you could clarify anything to a user or a potential customer about Visium HD data or the analysis process, what would you want to tell them?
Stephen: The first thing that I would tell someone is it might be scary, but it's also not impossible. The Visium HD analysis pipeline is honestly approachable for most people. You don't have to be a computer programmer or a bioinformatician to be able to look at things and get meaningful information.
The other thing that I would tell people is, although we have very fine resolution, this is not a single cell assay. Keep in mind when you're looking at your gene expression profiles on the bin level that most of these bins will have some sort of mixture of cells. It's not a pure population, but it's more pure than v2. And right now we don't do cell segmentation on Visium HD. That said, there are resources out there to do cell segmentation, or more likely nucleus segmentation, and really get closer to what an actual single cell is doing in space.
Advanced Visium HD analysis techniques and third party tools
This interview was conducted with Joey Arthur, PhD, Staff Computational Biologist at 10x Genomics. It was edited for length and clarity.
What are some third-party tools for Visium HD data analysis that you would recommend to customers and what do the tools do?
Joey: There are a few different categories of tools. There's at least one broad platform for spatial analysis called Squidpy which builds on the Scanpy ecosystem in Python. You could think of it like a scalable toolkit that is made up of many, many smaller tools and algorithms. It’s built from the ground up for spatial analysis and it has a lot of nice features. For example, it’s great for working with imaging data. With Visium HD, you have these images of the tissue that you put side by side or one on top of the other—the data on top of the image.
A lot of people in the single cell community use Seurat, which is in R, to do their analysis. Seurat also supports Visium HD and other spatial analyses, although in my experience it's easier to work with the imaging data in Python.
On top of that, there are a number of different tools for specific types of analysis that are unique to spatial. One of the big categories is tools for integrating single cell data with spatial data. You'll often hear these called cell type deconvolution tools. The two that I would recommend for Visium HD are Robust Cell Type Deconvolution (RCTD, implemented in the spacexr package) and Cell2Location. We have really high resolution with the Visium HD assay: the individual squares, they're very small—they're 2 µm x 2 µm, which is much smaller than a typical cell. But there's not a one-to-one match between squares and cells—like this square is exactly this cell, et cetera. So deconvolution tools are one thing that people will use to help their data be more interpretable in terms of known cell types. These tools are also useful because there is more and more single cell atlas data out there. People have collected lots of single cell datasets and labeled the cell types. Being able to apply that knowledge to a new spatial data set is really valuable.
[You can explore these and other community developed tools for deconvolution in our Analysis Guide.]
The other big class of tools is going to be cell segmentation. That’s where this Bin2Cell pipeline comes in that was presented in our webinar. I think that one is really great.
There's an R package called Voyager, and that has tools for doing what they call spatial statistics. There are a lot of statistical methods or data analysis methods that have been developed for geospatial data. Before the spatial transcriptomics revolution in the last so many years, most spatial data came from astronomical or geographical observations, and people have come up with a lot of different ways of thinking about that data. This Voyager package takes inspiration from that work and applies it to spatial-omics. They're working now to add support for Visium HD.
I would also call out that there are a lot of methods for finding spatially variable genes, meaning genes that have interesting spatial patterns and are not just distributed randomly. There are several tools for that within Squidpy and Seurat, as well as many other independently developed packages.
How do these third party tools complement the capabilities of Loupe Browser? How might you use them together?
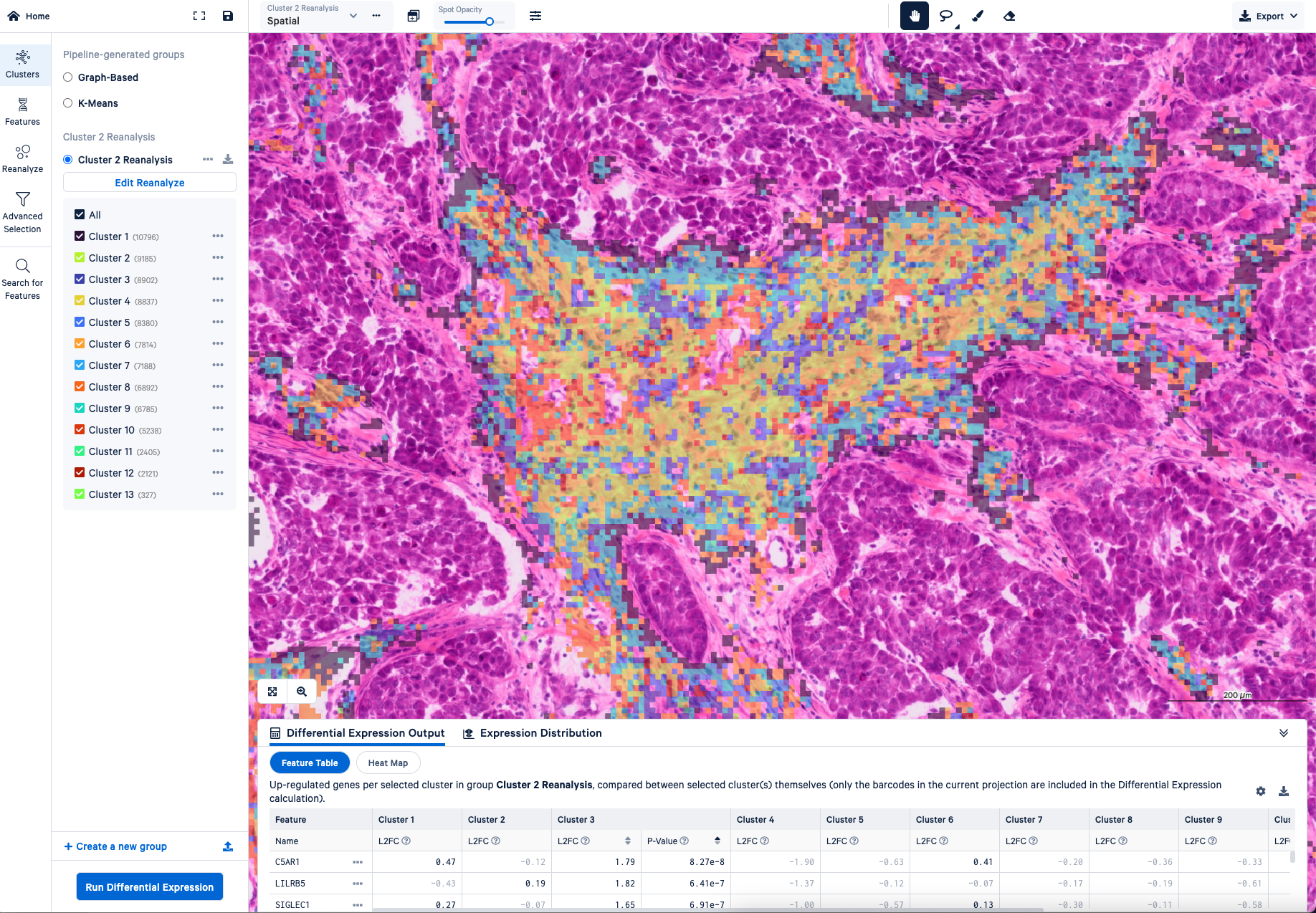
Joey: Loupe is great for letting you interactively explore the gene expression data and look at different genes or combinations of genes. You can really quickly scroll around the image, zoom in and zoom out, et cetera. It also allows you to perform the most essential analysis steps, like clustering the bins into groups and running differential expression to find marker genes. You can also filter out particular bins or dive deeper into a subset of the data using the Reanalyze tool.
Beyond that though is where, for the time being, you’ll have to go to third party tools. So more detailed spatial statistical analysis, that would be downstream tools. We don't have support yet for cell segmentation. There's aggregating data from single cell and spatial together, and looking at where the cell types are spatially—that also would need to be done in third party tools.
To me, Loupe is the best way to look at the raw data and get an initial look at things, because R and Python are just not quite as interactive.
What third-party capabilities are you most excited for and how do you think they could impact the kinds of discoveries customers can make from their HD data?
Joey: The biggest area of excitement for me is actually not covered by these tools. I’m excited about tools for comparing multiple samples and seeing how they're different. As these technologies are more adopted, I think we'll see large datasets of [samples from] patients with disease, or different subtypes of disease, and healthy controls. We want tools to characterize what's different between the healthy and the diseased samples—like asking how does the spatial localization of the cells change in these different groups? Those tools are not quite there yet. But I expect the ideal version of that kind of analysis to come in the next year or two probably.
One more type of analysis I find really interesting, which is implemented in Seurat and Squidpy, is spatial neighborhood analysis (sometimes called niche analysis). It's something beyond the usual clustering and differential expression workflow that we use for single cell, and it can only be done with spatial data. In some tissue contexts, the clustering results can be hard to interpret, but neighborhood analysis gives you much more interpretable spatial regions that match known morphology. Performing that on multiple samples and then looking at how the neighborhoods are different across different samples—I think that's a really interesting direction.
The equivalent could be like in single cell data, we look at differences in cell state. In this neighborhood analysis, it may be that the individual cells are quite similar in different situations, but they're sort of mixed together spatially in different ways. This unique composition produces a distinct spatial niche. So it could show you very quickly where immune hotspots are in a tumor, or the heterogeneity that you see there. I think it's a very good way of getting at if different parts of the tumor have different microenvironments.
Is there anything that we haven’t talked about that you would want to share with users about Visium HD data or the analysis process?
Joey: Another thing I'm pretty excited about is incorporating the imaging information more into the data analysis. One great feature of Visium HD is that it allows you to take an H&E or fluorescent image of the same section. We did a lot of work to make sure that people can overlay that imaging data on their gene expression data as accurately as possible. You have this really accurate alignment between the gene expression and the image that comes straight out of SpaceRanger, enabled by the Visium HD technology and the CytAssist instrument.
H&E images are really informative—pathologists use them to make all sorts of conclusions about health. So it should definitely be the case that combining the image with the gene expression data can get you even more insights into biology. Even at a basic level, you have to have that H&E image to do cell segmentation. If you don't have that, you can try to do segmentation on a serial section, but it won't be as good.
I think a lot of people are working on this. I would expect there to be some sort of deep learning or AI based system—machine learning methods for looking at the image together with gene expression data all at once. I think that's really exciting. And again, you can't do it at all with single cell, there's no image there. So it's kind of a new frontier for spatial.
Continue exploring resources for Visium HD data analysis:
- Webinar: Discover spatial insights with intuitive analysis tools for Visium HD data (featuring Bin2Cell and SpatialData)
- Software analysis support for SpaceRanger and Loupe Browser
- Visium HD datasets
- Analysis Guide: Nuclei Segmentation and Custom Binning of Visium HD Gene Expression Data
Blog contributors: We’d like to thank Stephen and Joey for providing their expert insights!

