The cellranger multi pipeline is used to analyze Universal 3' and 5' Gene Expression, V(D)J, Flex v1/v2, and Feature Barcode (CRISPR Guide Capture, Antibody Capture, and Antigen Capture) data. It can be used for both singleplex and multiplex expereiments.
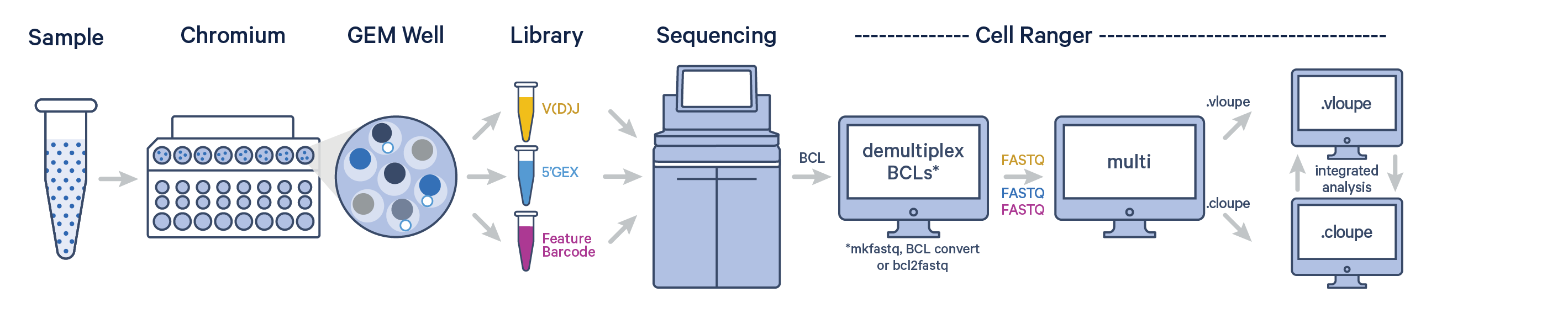
The multi pipeline takes a config CSV with paths to FASTQ files derived from Gene Expression, Feature Barcode, and V(D)J libraries originating from a single GEM well. It performs several functions, including alignment, filtering, barcode counting, and UMI counting. It also performs sequence assembly and paired clonotype calling on the V(D)J libraries. Additionally, the cell calls provided by the gene expression data are used to improve the cell calls from the V(D)J data. Using cell barcodes, multi generates feature-barcode matrices, identifies clusters, conducts gene expression analysis, and provides preliminary cell type annotations.
This pipeline is recommended for analyzing combined 5' Gene Expression and V(D)J libraries, with or without Feature Barcode libraries, from the same sample. Additionally, it is the only available pipeline for analyzing 3'/5' Sample Multiplexing, Flex v1/v2, and 5' Antigen Capture (BEAM) data.
Visit the multi tutorial page for self-guided and video tutorials on running cellranger multi.
Use the pipeline selector tool to identify a suitable pipeline for your experimental design.
The cellranger multi pipeline is required to analyze samples multiplexed using on-chip multiplexing (OCM), Antibody Capture, or 3' CellPlex.
For singleplex libraries containing Gene Expression and V(D)J data (with or without Feature Barcodes), cellranger multi is the recommended pipeline, as it refines V(D)J cell calls using gene expression data (described below).
The cellranger multi pipeline improves cell calls in the V(D)J dataset
by discarding any cells that were not also called in the corresponding 5'
Gene Expression dataset. By assigning cells that are called in the V(D)J
results but not in the 5' Gene Expression results as background GEMs in
the V(D)J data, cellranger multi mitigates any overcalling issues that
may arise in V(D)J data. This improved cell calling is only possible
when both 5' Gene Expression and V(D)J libraries were sequenced from the
same sample.
As shown in the image below, final V(D)J cell calls (intersection area) exclude cells that were only called by the vdj pipeline (yellow region).
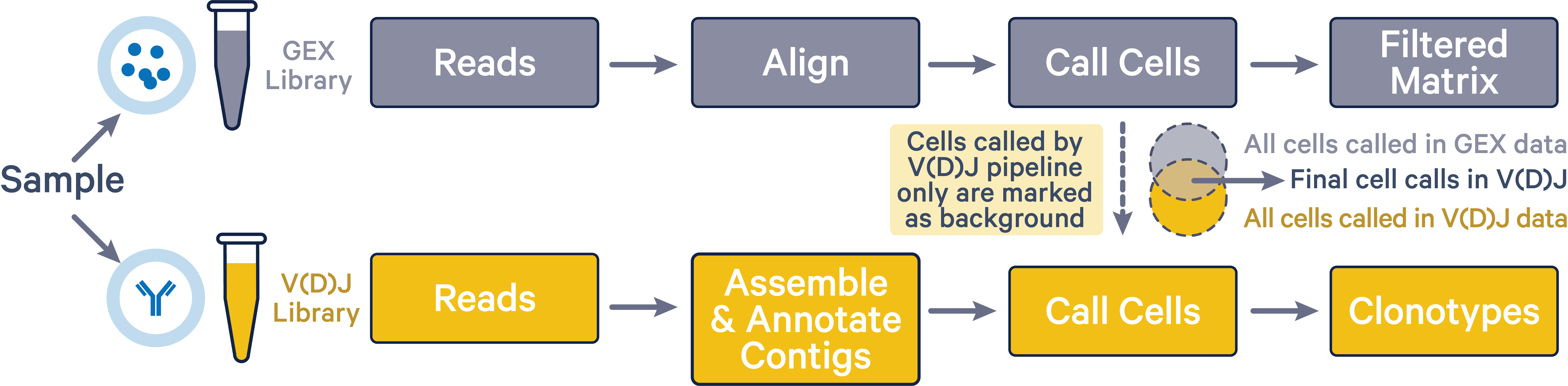
The 5' Gene Expression cell calls are not affected by the cellranger multi pipeline. The Gene Expression library is representative of the entire pool of poly-adenylated mRNA transcripts captured within each GEM. VDJ-T or VDJ-B transcripts within the Gene Expression library are then selectively amplified to create the V(D)J library. As a result, the Gene Expression library has greater sensitivity in detecting GEMs that have cells compared to the V(D)J library. When the cellranger multi pipeline is executed with both 5' Gene Expression and V(D)J data, any barcodes that are not classified as cells in the 5' Gene Expression data are removed from the V(D)J cell set. This process ensures that only the barcodes identified as cells in the Gene Expression library are retained in the V(D)J library for downstream analysis.
Visit the List of Inputs page for a comprehensive list of all inputs required to run the Cell Ranger multi pipeline.
The cellranger multi pipeline takes a config CSV file as input. The config CSV contains paths to FASTQ files for any combination of V(D)J, Gene Expression, and/or Feature Barcode libraries. Go to the Cell Ranger multi config CSV page for a complete list of options for each section.
To generate FASTQ files, use a demultiplexing tool.
To simultaneously generate single cell feature counts, V(D)J sequences, and annotations for a single library, run cellranger multi with the following arguments:
| Argument | Description |
|---|---|
--id | A unique run ID string: e.g. sample345 that is also the output folder name. Cannot be more than 64 characters. |
--csv | Path to multi config CSV file enumerating input libraries and analysis parameters. |
The multi config CSV contains both the library definitions and experiment configuration variables. It is composed of up to four sections: [gene-expression], [feature], [vdj], [antigen-specificity] and [libraries].
The [gene-expression], [feature], [vdj], and [antigen-specificity] sections have at most two columns and are responsible for configuring their respective portions of the experiment. The [libraries] section specifies where input FASTQ files may be found.
Starting with Cell Ranger v9.0, you can enable automated cell type annotations in a multi run by adding parameters to the [gene-expression] section of your multi config CSV. See the example multi config CSV below. Please note that automated cell type annotations are available only if a Gene Expression library is included in your multi analysis. The currently available annotation models are in beta.
Example multi config CSVs can be downloaded from public datasets. Cell Ranger v7.1 and later also provides the option to download a multi config CSV template via the command line.
Generate a multi config CSV template by running cellranger multi-template, see usage here.
After determining the input arguments, run cellranger multi. Remember to
customize the code with your sample id and csv file path:
mkdir /home/jdoe/runs
cd /home/jdoe/runs
cellranger multi --id=sample345 --csv=/home/jdoe/sample345.csv
Following a series of checks to validate input arguments, cellranger multi pipeline stages will begin to run:
Martian Runtime - v4.0.8
Running preflight checks (please wait)...
...
By default, Cell Ranger will use all of the cores available on your system to execute pipeline stages. You can specify a different number of cores to use with the --localcores option; for example, --localcores=16 will limit Cell Ranger to using up to sixteen cores at once. Similarly, --localmem will restrict the amount of memory (in GB) used by Cell Ranger.
The pipeline will create a new folder named with the run ID you specified using the --id argument (e.g. /home/jdoe/runs/sample345) for its output. If this folder already exists, Cell Ranger will assume it is an existing pipestance and attempt to resume running it. If you wish to re-start the run, delete the output folder (sample345/ in this example) and rerun the pipeline.
A successful cellranger multi run should conclude with a message similar to this:
Waiting 6 seconds for UI to do final refresh.
Pipestance completed successfully!
yyyy-mm-dd hh:mm:ss Shutting down.
Saving pipestance info to "tiny/tiny.mri.tgz"
To learn more about the output files generated, refer to the Outputs for multi section.
The cellranger multi pipeline supports downsampling the reads by specifying a rate between 0 and 1 independently for each library. It also allows trimming the reads to a fixed length, which is not supported in the cellranger vdj pipeline.