Searching for developmental mechanisms of lung pathology with a single cell and spatial atlas of human fetal lung

Over 100,000 individuals globally are living with cystic fibrosis (1). Most are diagnosed by age 2. While scientists have identified the rare cell population that plays a key role in cystic fibrosis biology, there’s still a lot to learn about the underlying developmental errors and local cellular relationships that may give rise to or exacerbate this life-changing disease.
Cystic fibrosis is one of a number of genetic diseases that primarily cause lung pathology, including familial pulmonary fibrosis or surfactant metabolism dysfunction (2). Structural or functional lung abnormalities can also develop in a fetus during pregnancy. Therefore, deeply characterizing the healthy human lung, particularly in the earliest stages of fetal development, can be an effective approach to understand the cellular origins of these lung disorders as well as the impact of disease on lung function.
Motivated to establish an atlas of healthy human fetal lung development, a team of researchers from the Hospital for Sick Children in Toronto and the University of Toronto leveraged high-resolution single cell and spatial technologies to profile the cellular composition and spatial organization of human fetal lung tissue from 10 to 19 gestational weeks (3)—a significant work also due to the precious nature of the human samples, as most lung development studies have previously leveraged mouse tissue. This research represents an important step towards defining the cellular and molecular mechanisms of lung diseases like cystic fibrosis and reflects on the huge potential of combining high-resolution single cell and spatial approaches to better understand the development of complex tissue systems.
Building a single cell atlas of human fetal lung yields questions regarding developmental origins of important cell types
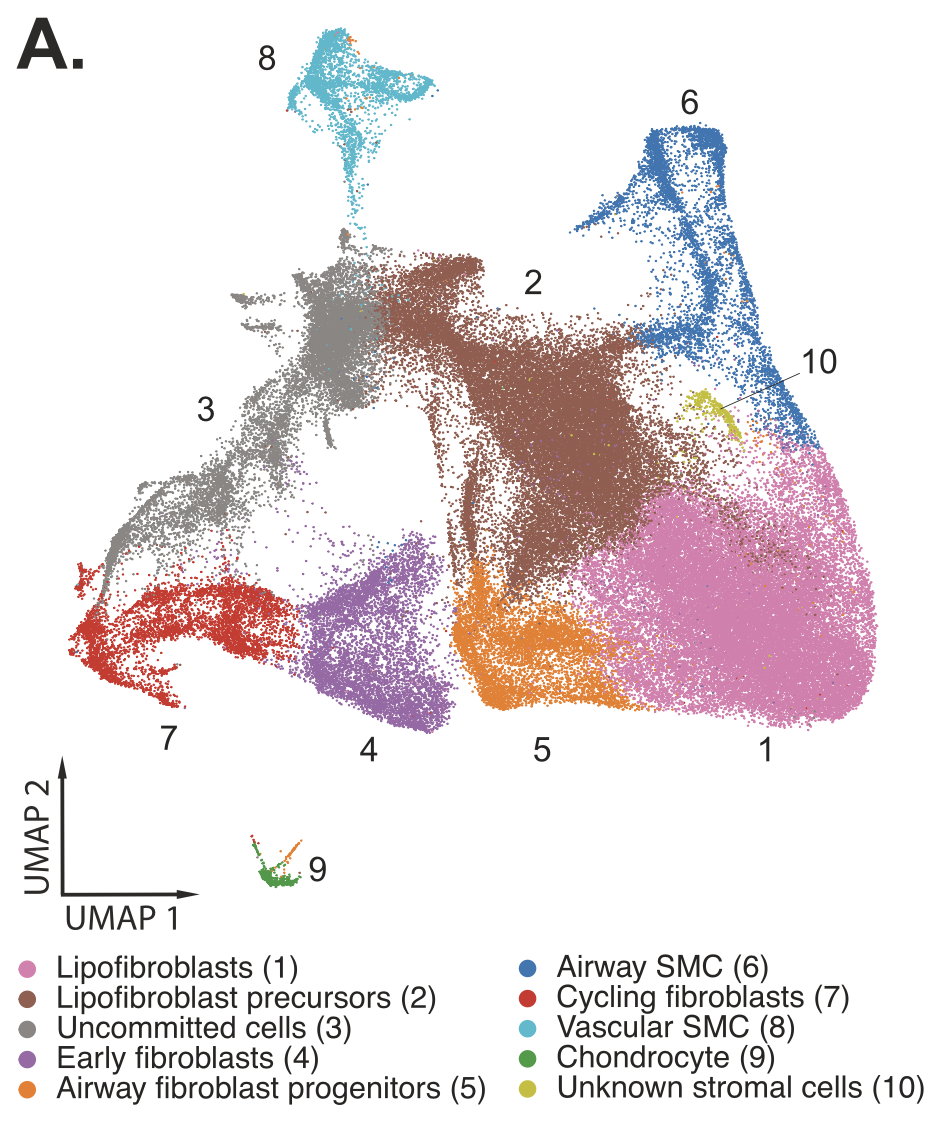
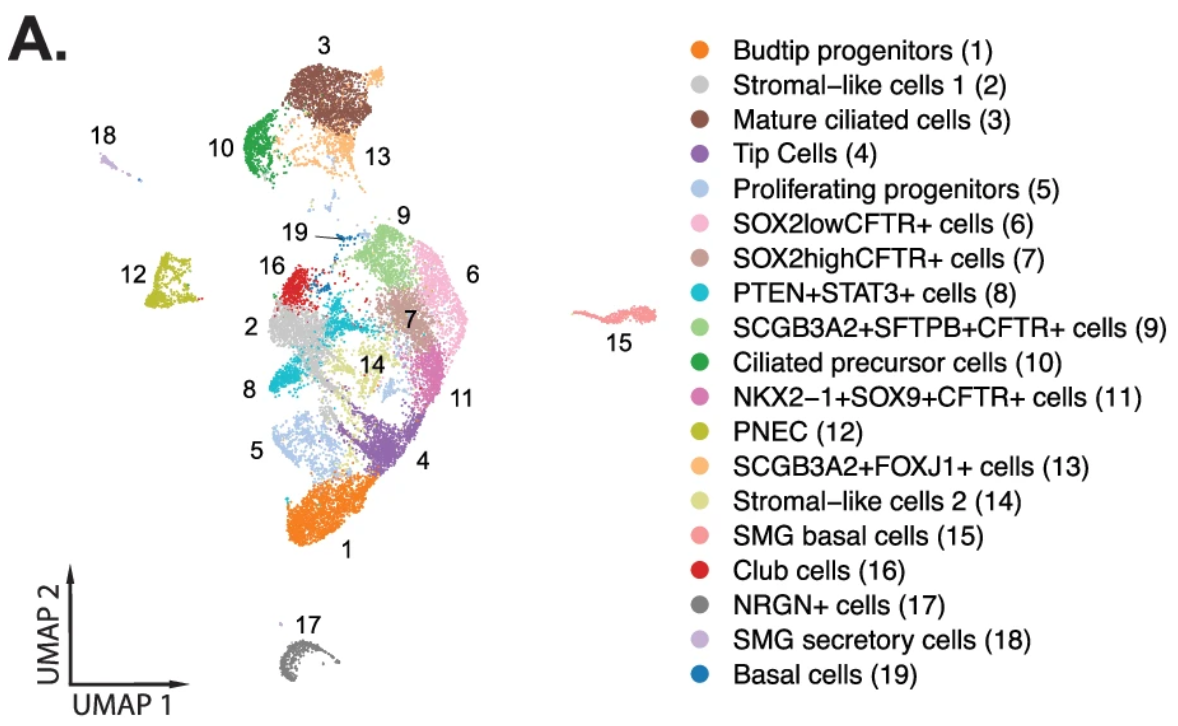
Understanding the detailed cellular composition of the human fetal lung was the first step for the team, leading them to use Chromium Single Cell Gene Expression to profile fresh tissues taken from 19 fetal lung samples spanning 10–19 gestational weeks. From 156,698 total cells analyzed, they identified 58 distinct cell types and states (Figure 1), of which stromal cells were the largest cell class proportionally (Figure 2,3). Stromal cells play diverse roles in the lung— like modulating immune responses, as well as damage and repair processes—and can even differentiate into myofibroblasts that contribute to fibrotic lung pathology (4). These roles and the possibility of disease association make stromal cells an important cell class to evaluate in a developmental context.
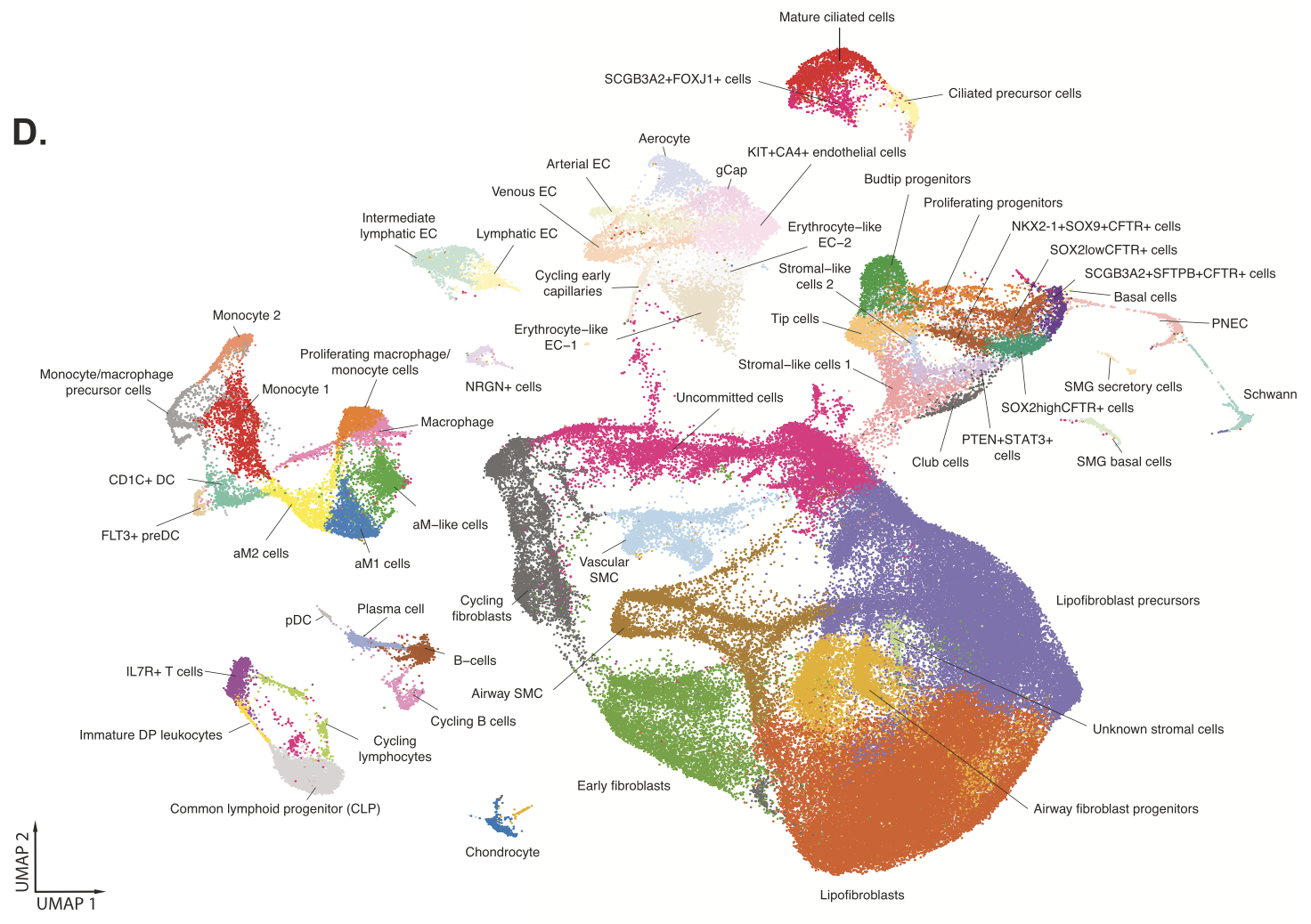
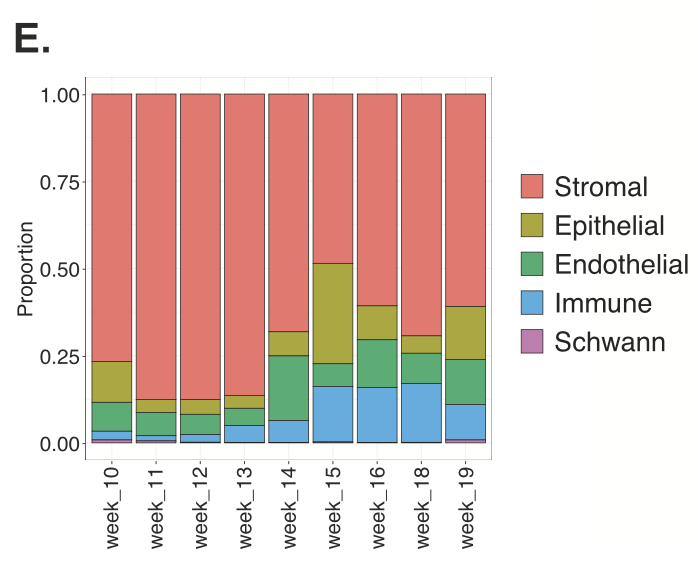
Comparing this fetal lung single cell dataset to an adult lung single cell atlas using the Seurat MapQuery function (which allowed them to identify cells of similar biological states based on a prediction score), the team noted that 83% of fetal lung cells were captured in the adult dataset. But clearly there were many cells that did not overlap. For example, NKX2-1+ SOX9+ CFTR+ fetal lung cells—a population of epithelial progenitor cells that have the potential to differentiate into various epithelial cell types, including cells that are crucial for the branching structure of the lung (3)—were not seen in the adult dataset, emphasizing the role this cell plays in the gestational time period the fetal tissues sampled. Reversing the analysis, the team observed that only 56% of adult lung cells were captured in the fetal dataset. Significantly, cells that were not observed in the fetal dataset included alveolar fibroblasts and macrophages, nasal cells, and ionocytes, the latter of which is a rare cell type that is a known contributor to cystic fibrosis biology.
This comparison analysis yields a number of questions: How could the contrasting cellular composition of fetal and adult lung tissue affect the onset of developmental diseases? When do the cells that possibly contribute to disease arise in development?
These results also suggested that a complementary approach to fully uncover the cellular composition of the sample would be valuable, particularly if that approach allowed a researcher to see comprehensive cell states undisturbed in their native context. This is a need that other research groups have commented on. According to Mayr et al., “Spatial transcriptomics addresses this by providing spatially resolved gene expression within the natural tissue environment of intact tissue without inducing cell stress, death, or an isolation bias” (5).
Adding spatial context to map epithelial–stromal cell relationships in the human fetal lung
Seeking to add more clarity to their single cell findings about human fetal lung composition and to place those cell types in their appropriate spatial context, the team turned to Xenium In Situ single cell spatial imaging. Using the 289-gene Human Lung Gene Expression Panel, supplemented with a 50 custom gene add-on panel derived from their single cell dataset, the team performed Xenium In Situ analysis on two archived FFPE human fetal lung samples taken from gestational weeks 15 and 18.
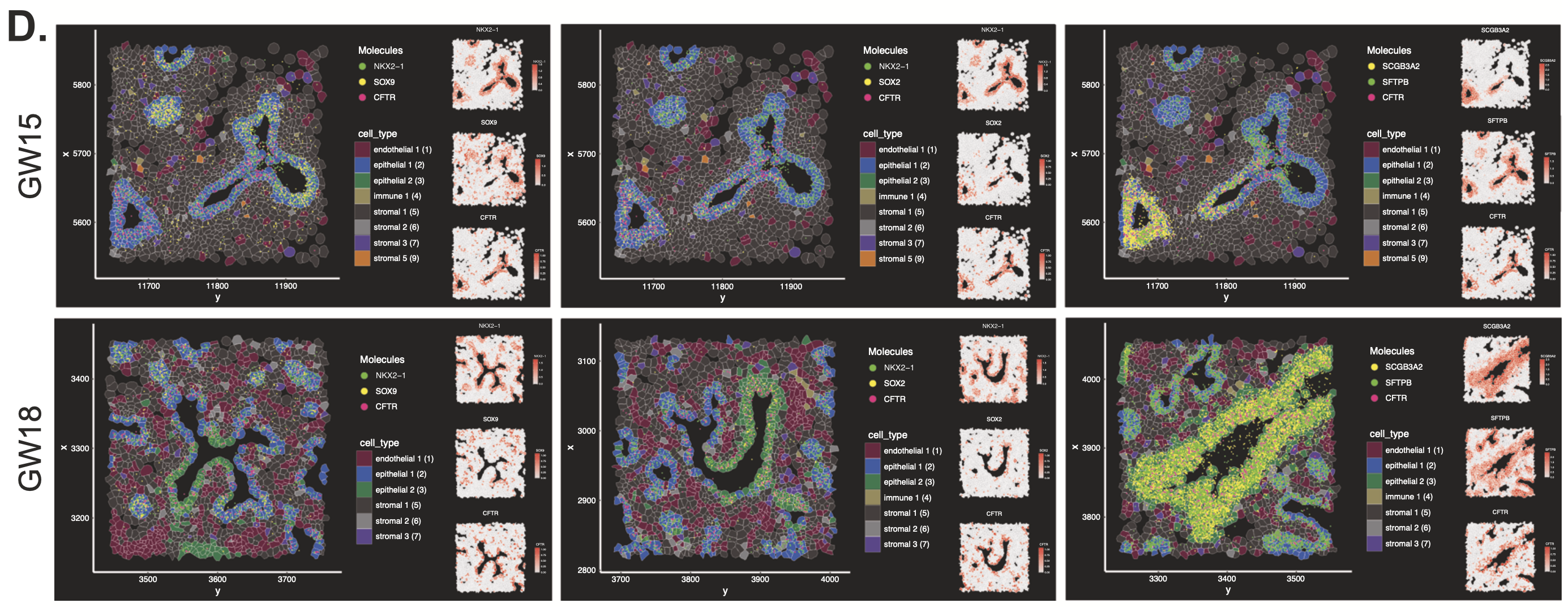
Clustering cell populations by differentially expressed genes (DEGs), they identified 9 major cell clusters in GW15 tissue and 7 major clusters in GW18 tissue. Those clusters were mapped spatially onto the tissue sections to visualize their regional distribution. Xenium data was also used to spatially localize the distribution of some of the DEGs from 10 stromal cell subtypes that had been identified in the single cell dataset, revealing how stromal subtypes like fibroblast progenitors colocalized with epithelial subtypes that compose important structural regions of the lung tissue, such as the large airways (Figure 4).
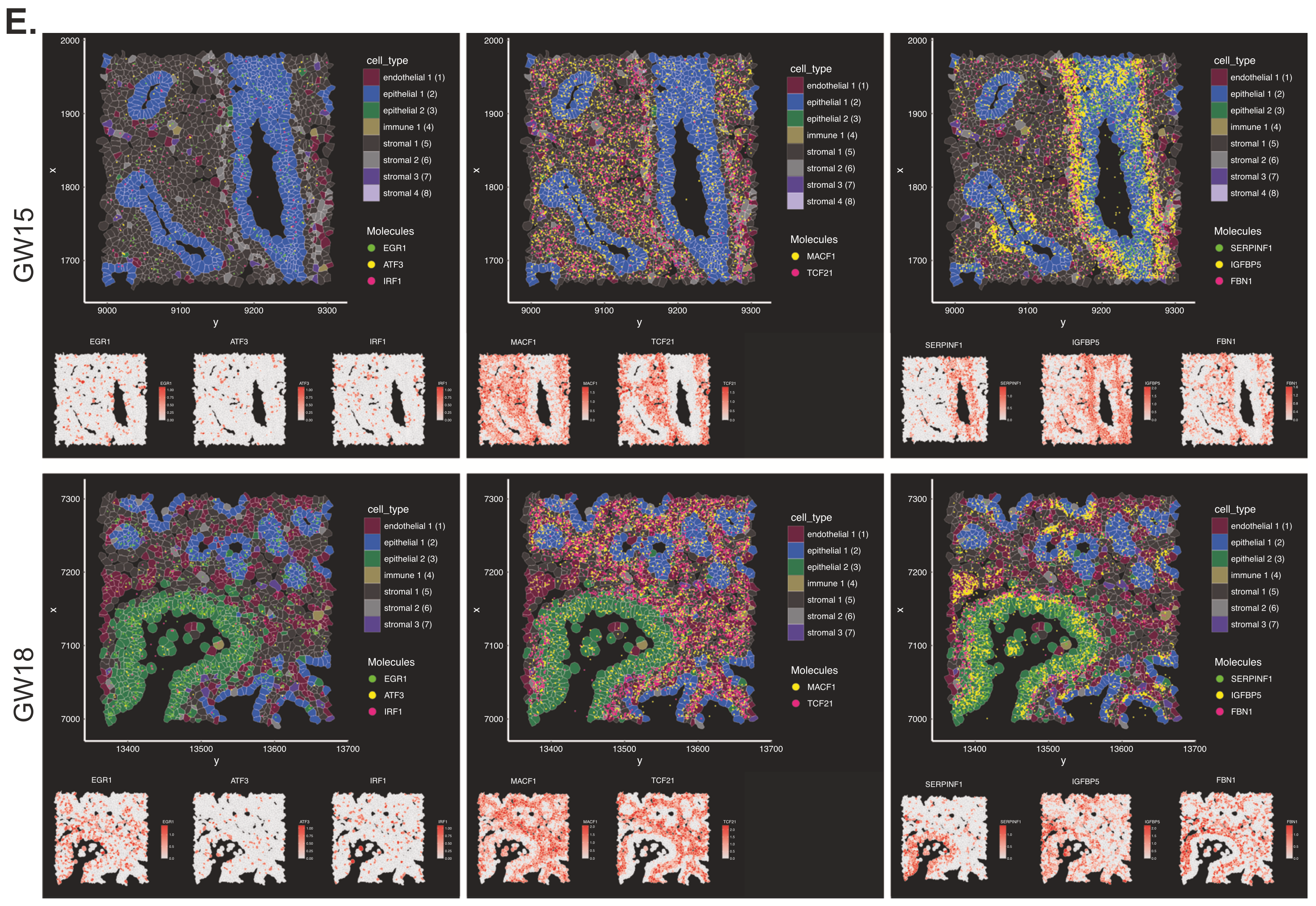
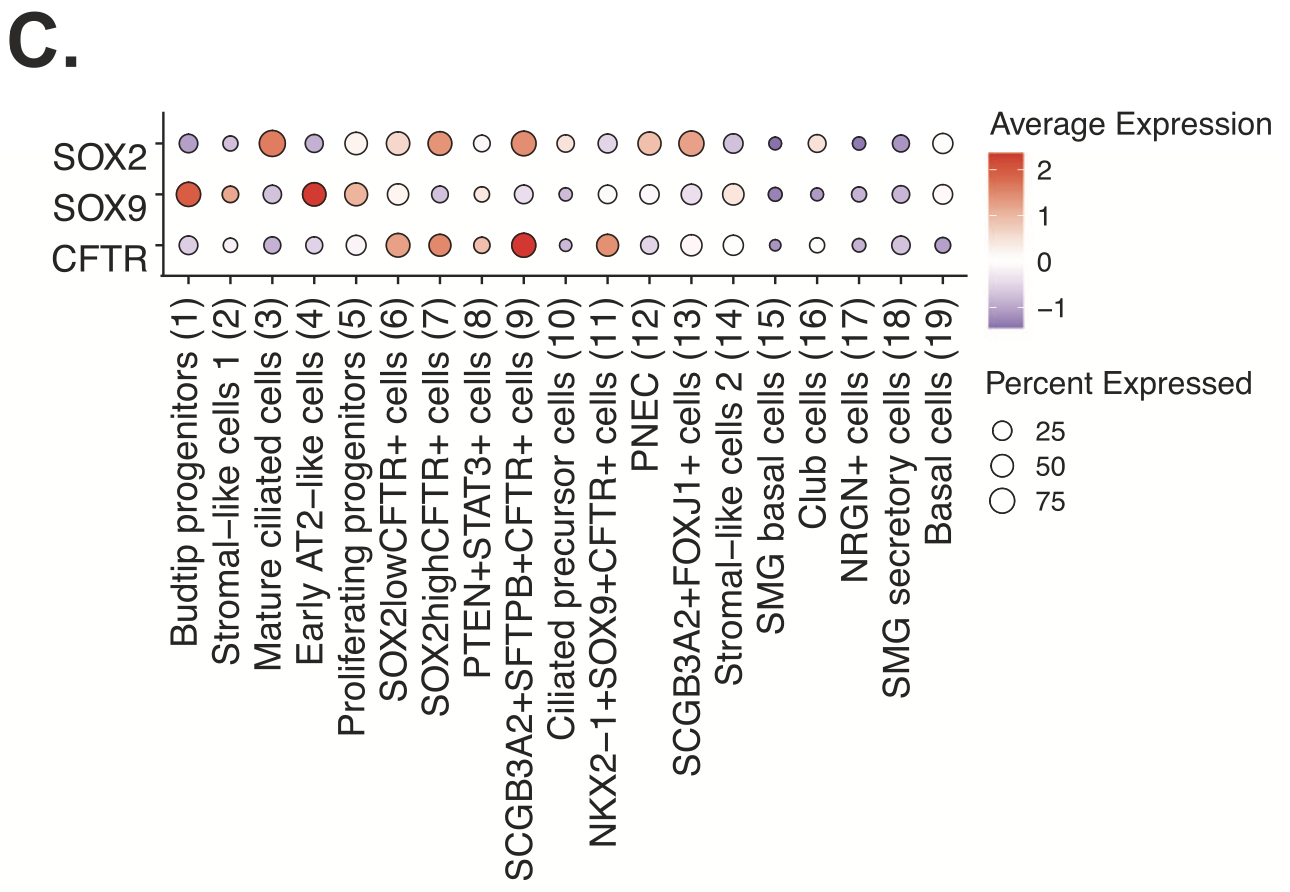
An evaluation of epithelial cell diversity based on single cell RNA-seq data also revealed 19 unique subtypes, four of which expressed higher levels of CFTR (cystic fibrosis transmembrane conductance regulator; Figure 5,6). Mutations in CFTR cause cystic fibrosis, but the gene may also play a role in lung branching morphogenesis. Xenium analysis of the same two tissue sections (Figure 7) demonstrated that, of these four epithelial cell types, one population (NKX2-1+ SOX9+ CFTR+) was restricted to regions where lung tissue was beginning to branch. Another population, SOX2+ CFTR+, was restricted to stalk regions of the airways. Given the known proliferative role of SOX2, the team hypothesized SOX2+ CFTR+ cells are involved in driving expansion of the epithelial cells that form the developing airways.
Going deeper into cell lineage and signaling relationships with single cell and spatial transcriptomics
Expanding their investigation to explore the identities and relative localization of additional epithelial cell subtypes, the team leveraged Visium Spatial whole transcriptome analysis on two additional FFPE tissue sections from the same archived GW15 and GW18 fetal lung samples. Visium whole transcriptome spatial data paired with Xenium data allowed them to assess shared gene expression between cell populations that could give insights into their lineage relationships. For example, they saw that tip cells and bud tip progenitor cells both expressed common genes, including some related to surfactant protein production (an interesting finding in that surfactant metabolic dysfunction is associated with genetic lung diseases that can cause neonatal respiratory failure (2)). They also observed that bud tip progenitors and tips cells were spatially correlated—analysis of spatial transcriptomics data suggested they were likely colocalized within the same Visium spots—and therefore could represent the same cell type, just in different phenotypic states.
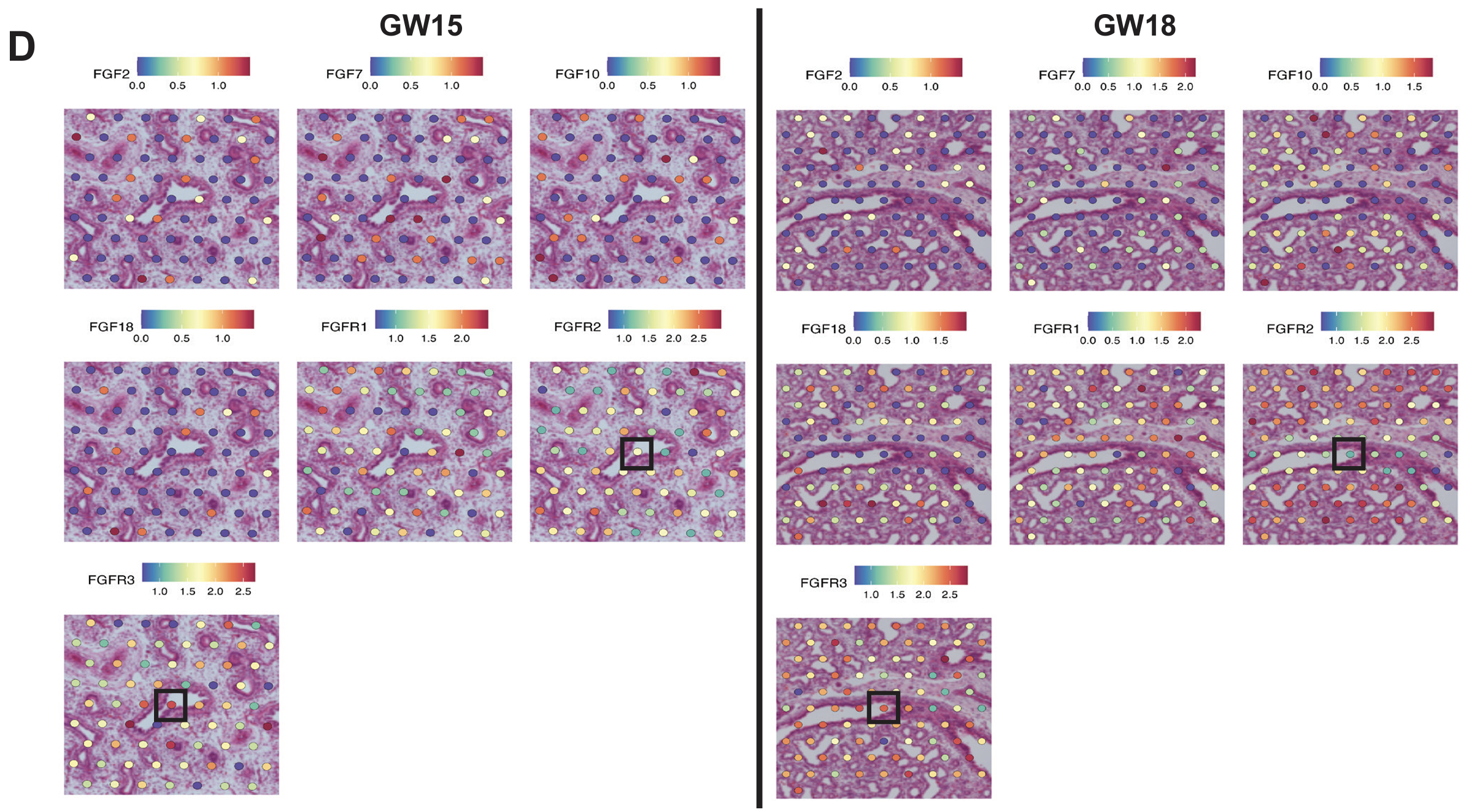
The team also leveraged their single cell RNA-seq and Visium spatial transcriptomics datasets to explore possible ligand–receptor interactions between fetal epithelial and stromal populations with CellChat, a software that analyzes and infers cell-to-cell communication networks based on single cell annotations and spatially close cell types (determined by Visium data). This allowed them to identify signaling pathways targeting a triple-positive CFTR-expressing epithelial cell population of interest (SCGB3A2+ SFTPB+ CFTR+), including a fibroblast growth factor (FGF) signaling pathway that is likely involved in lung branching morphogenesis. Single cell and Visium data demonstrated heightened expression of FGF receptors directly in these triple-positive cells or in tissue regions with a high proportion of the triple-positive cell population (Figure 8), suggesting a signaling relationship between airway fibroblast progenitors and this triple-positive cell type.
Improving development of hPSC-derived fetal lung organoids with single cell data
A secondary goal of the study was to define developmental processes in primary human fetal lung tissue that could improve differentiation protocols for lung cells and fetal lung organoids derived from human pluripotent stem cells (hPSCs), as well as better define the developmental stage of those cultured cells. To achieve this, the team performed single cell RNA-sequencing on established hPSC-derived fetal lung cells and fetal lung organoids. Then they compared DEGs from hPSC cell clusters to those found in the primary fetal lung tissue to identify the cell types in the cultures or organoids that overlap with those present in the primary tissues and align them to a developmental stage. Their analysis revealed that hPSC-derived fetal lung cells correlated to lung epithelial tissue younger than GW12, while hPSC-derived fetal lung organoids correlated to lung epithelial tissue older than GW16. Both the cultured cells and organoids were also shown to be distinct from adult lung composition, confirming their utility for future studies.
The value of three modalities to study fetal lung development
Development, like all of biology, is far from straightforward. One study may reveal more questions or provide only a partial view of the true complexity of a developing organ. As in this case, the research team acknowledged that “the rare ionocytes that express the most abundant CFTR transcripts in postnatal lung tissues were not detected in any of the fetal lung tissues examined. However, this does not preclude the development of these cells later in fetal lung development.” This “non-discovery” is an important result in and of itself, suggesting these cell types, crucial to the underlying biology of cystic fibrosis, likely arise at a later gestational stage.
Together, high-resolution single cell and spatial tools can provide this deeper understanding of the cellular composition and spatial organization of developing organs over time, helping scientists combine data pieces to form hypotheses around cellular functions and lineage relationships that may inform our future understanding of the cellular and molecular mechanisms underlying developmental diseases.
In this study, specifically, Chromium Single Cell Gene Expression was able to give:
- Broad oversight to the cellular composition of the human fetal lung, parsing out major cell classes and refining detailed cell states with whole transcriptome readouts and analysis of DEGs
- Strategic guidance regarding gene content to include in a targeted panel for single cell spatial imaging
Xenium In Situ provided:
- A refined view of single cells, including diverse epithelial and stromal cell subtypes, in native tissue context, ensuring complete representation of the cell types present in the tissue
- Precise localization of key transcripts/DEGs across the tissue section to inform cell-type identification
- Visualization of local cellular relationships and possible associations with structural processes in the developing airways
And Visium Spatial Gene Expression empowered:
- Unbiased analysis of shared gene expression programs between colocalized cell populations to infer their identities, lineage relationships, and possible cell-to-cell communication networks
See the data for yourself by reading the publication! And explore another application of all three 10x Genomics single cell and spatial platforms to human colon cancer in our recent bioRxiv preprint.
References:
- https://www.nhlbi.nih.gov/health/cystic-fibrosis
- Devine M and Garcia C. Genetic interstitial lung disease. Clin Chest Med 33: 95–110 (2012). doi: 10.1016/j.ccm.2011.11.001
- Quach H, et al. Early human fetal lung atlas reveals the temporal dynamics of epithelial cell plasticity. Nat Commun 15: 5898 (2024). doi: 10.1038/s41467-024-50281-5
- Doherty D, et al. The role of lung resident mesenchymal stromal cells in the pathogenesis and repair of chronic lung disease. Stem Cells 41: 431–443 (2023). doi: 10.1093/stmcls/sxad014
- Mayr C, et al. Spatial transcriptomic characterization of pathologic niches in IPF. Sci Adv 10: eadl5473 (2024). doi: 10.1126/sciadv.adl5473
About the author:
