Avoiding that agitated feeling when studying sub-populations: scRNA-seq with CRISPRi-perturbed cells

Dr. Britt Adamson, a Postdoctoral Fellow from the Weissman Lab at UCSF, spoke at our 2017 Bay Area User Group Meeting about her work tackling the dynamic transcriptional mechanisms of the unfolded protein response (UPR) and how they were affected with various input stresses. She and her colleagues systematically dissected the complex UPR stress-response pathway using a combination of CRISPR interference (CRISPRi) perturbations and single cell RNA-seq analysis to simultaneously identify the introduced perturbations and measure their transcriptional impact on a cell-by-cell basis.
The UPR is a complex stress-response pathway
The accumulation of unfolded proteins and the resulting increase in endoplasmic reticulum (ER) stress is observed in numerous diseases such as cancer, autoimmune disease, and neurodegenerative disorders, to name a few.1 Because of this, therapies targeting various components of the UPR for the intervention and treatment of human disorders are especially appealing.2 The UPR is generally deployed in response to ER stress catalyzed by misfolded proteins, but can also be activated by developmental cues, calcium dysregulation, and other cellular stresses. In response to stress three ER-transmembrane sensors (IRE1ɑ,ATF6, and PERK) activate transcription factors that promote cell survival or, when the ER stress cannot be corrected, promote cell death.
In an attempt to greater understand the UPR, Britt wanted to measure CRISPRi-mediated perturbation UPR transcriptional responses at the genome-scale. However, it was crucial that she was able to tease out the responses to these perturbations on a cell-by-cell basis. In this case, traditional bulk RNA sequencing methods would not allow for the detection of subtle differences in single-cell transcriptional responses because of the limitation that only an average transcriptional response of all the cells receiving a particular perturbation could be measured using that method. In order for her screen to be successful, Dr. Adamson needed to build a platform in which she could scale the amount of CRISPRi-mediated perturbations, while also measuring single-cell transcription in order to document perturbation responses—in came the development of the Perturb-seq Platform.
The Perturb-seq Platform
The CRISPRi-mediated perturbations allowed for sequence-specific repression of gene expression at the transcriptional level, leading to the fine-tuning of various response mecha!
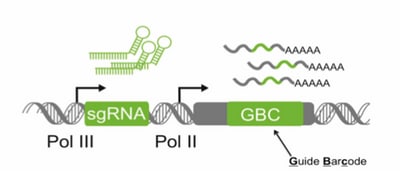{kind=link}
The Perturb-seq guide capture design: maintains fidelity of sgRNA and GBC pairing, ensures robust sgRNA activity and GBC expression, and ensures good sequencing coverage of identity-encoding transcript (GBC).nisms. These perturbations were introduced with lentiviral pools of small guide RNAs that allowed Britt to map the cell, via transcriptional read-outs, to its particular perturbation. To enable identification of the cell specific perturbations alongside the cellular transcripts, she designed an expression construct in which a Pol II transcribed guide barcode (GBC) is co-expressed with the Pol III transcribed sgRNA. Because the GBCs are transcribed by Pol II, the RNAs are polyadenylated, thus allowing them to be easily captured on the 10x Genomics Chromium™ System.
Powerful Possibilities with Perturb-seq
As mentioned above, studying the stress response can be tricky, but the Perturb-seq platform comes with some powerful potential. One way Britt used it was to identify which transcriptional regulons were specific to the signaling sensor molecules involved in the execution of the UPR. She performed an experiment to knockdown these molecules individually or in paired combinations, built the Perturb-seq cell library, and then exposed the cells to pharmacological agents that cause ER stress. Perturb-seq was then performed using the Chromium System.
She was able to map the activation of stress responses through the loss of biological systems in the cell: depending on the stress, there was a heterogeneous activation of these main signaling branches. For example, there were clusters of genes that were involved with PERK and IRE1 activation, but that were not involved in the activation of ATF6. Conversely, another set of genes were identified that activated ATF6 but not PERK.
She also questioned whether single cells can execute a heterogeneous response to a homogeneous perturbation.

Single cell RNA-seq reveals two subpopulations of BiP (HSPA5) depleted cells. Despite having the same BiP depletion, the celluar subpopulations elicit two different UPR pathway responses.depleted of BiP (a major ER chaperone, gene name HSPA5) where all three branches of the UPR are activated, were examined. Surprisingly, she demonstrated that the BiP depleted cell populations had very different patterns of gene expression. Using a custom analytical pipeline to compare control v. BiP depleted cells, the researchers identified independent components in the datasets. One component in particular bifurcated the BiP depleted cells, showing that different UPR branches were activated in different ways in cells containing the same BiP perturbation. Transcriptional data showed no difference in the level of BiP knockdown in the two cell populations further indicating that this was indeed a heterogeneous response to the same perturbation.
The team’s work demonstrates the utility of Perturb-seq for high-throughput functional genomics studies and the power of this approach to dramatically increase our understanding of complex, overlapping biological pathways. If you would like to read more in-depth information about this project, please refer to her elegant manuscript published in Cell last year or watch Britt’s video presentation on this exciting work here.
Single-Cell RNA-seq as a Tool for Studying Stress Responses
These findings illustrated the disparate ways in which the UPR can be executed, even in a seemingly homogeneous cell population. In fact, although these cells were subjected to the same stress, they responded in very distinct ways. Bulk RNA-seq would have described a state in which none of the cells actually existed. The utility of single-cell RNA-seq using the Chromium System was most apparent, however, when Britt and her colleagues observed the marked behavioral differences in BiP-depleted cells: all of these cells had the same genetic depletion, but the differences in individual cell responses could be clearly mapped. These results then bring into question what the causes of such differences are, and will open up a wide avenue of future exploration.
Additional Resources
- Read the Perturb-seq paper here.
- Learn more about the Chromium™ Single Cell 3' Solution.
- Read Chromium™ Single Cell 3' Solution application notes.
- Check out our data analysis and visualization tools - Cell Ranger™ and Loupe™ Cell Browser.
- Download Chromium™ Single Cell 3' Solution datasets.
References
-
Wang, S and Kaufman, R.J., "The impact of the unfolded protein response on human disease," J Cell Biol. 2012 197(2012): 857–867.
-
Hetz, C., Chevet, E., Harding, H.P., "Targeting the unfolded protein response in disease", Nat Rev Drug Discov 12 (2013): 703-19.